【トピックス】
デオキシヌクレオシドの新製法開発
及川利洋、小松弘典、石橋大樹
三井化学・触媒科学研、三井化学・触媒科学研、三井化学・生産技術研
1.はじめに
4種のデオキシヌクレオシド、デオキシアデノシン (dA)、デオキシグアノシン (dG)、デオキシシチジン (dC)、チミジン (T) は、遺伝子 (DNA) を構成する化合物である (図1)。
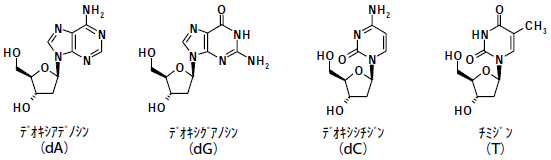
図1 2’-デオキシヌクレオシドの構造式
これまでは、エイズ薬アジドチミジン (AZT) の原料であるチミジンを除き産業的用途は必ずしも大きくなかったが、近年DNAチップなどの遺伝子診断用途や、アンチセンスなどの遺伝子医薬の製造原料として、新たな利用価値が見出され需要が拡大しつつある1)。
これまでの製造法は、鮭の白子から粗DNAを抽出し、加水分解後に各デオキシヌクレオシドを精製単離する方法であった。およそ100 tの鮭から、4種のデオキシヌクレオシドを約55 kg得ることができるとされている2)。この方法では今後の需要増大に対応できないだけでなく、コストダウンにも限界があった。
デオキシヌクレオシドの化学合成法は数多く報告されているが、チミジン以外では、コスト的に見合う方法はなかった。
酵素合成法としては、ヌクレオシドホスホリラーゼの正反応および逆反応を利用して、核酸塩基を交換し種々のデオキシヌクレオシドを合成する方法や (図2A)3)、ヌクレオシド-デオキシリボシルトランスフェラーゼ (EC 2.4.2.6) を利用し、核酸塩基を交換することで種々のデオキシヌクレオシドを合成する方法が知られていた (図2B)4)。しかし、デオキシヌクレオシドを原料とし他のデオキシヌクレオシドを合成する方法では、コスト的な問題に加え他のデオキシヌクレオシドの混入という品質的問題があった。他方、ヌクレオシドホスホリラーゼの直接的な原料である、デオキシリボース-1-リン酸 (dRP) を、工業的に化学合成することは極めて困難であった。
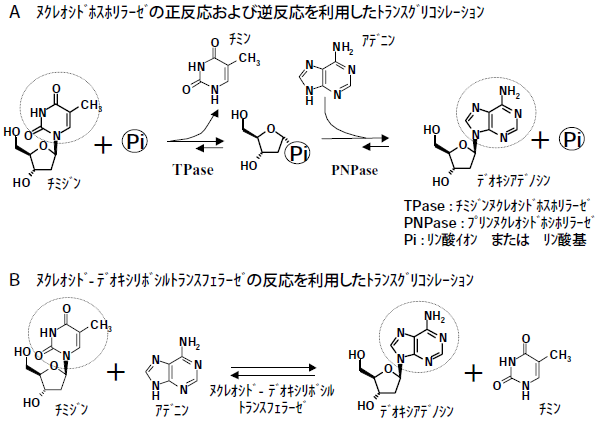
図2 デオキシヌクレオシドの酵素合成法
このようなことから、新しい発想によるデオキシヌクレオシドの製法開発が必要と考え、dRPの化学合成法とヌクレシドホスホリラーゼによるdRPのグリコシル化反応を組み合わせた、Chemo-Enzymaticな製法の開発に取り組んできた。その結果、4種すべてのデオキシヌクレオシドを共通のプロセスで合成する製法を開発することに成功した (図3)5)。本稿では、これらの製造法開発について紹介する。
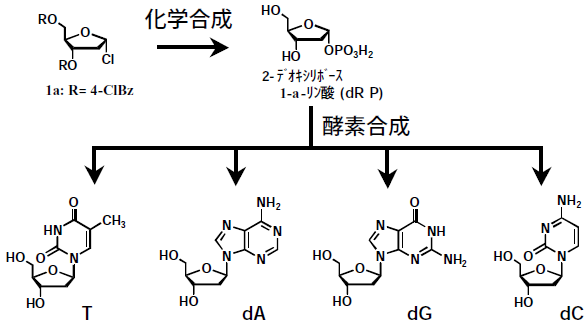
図3 Chemo-Enzymaticなデオキシヌクレオシド合成スキーム
2.デオキシリボース-1-リン酸 (dRP) 合成法の開発
クロロ糖 (図4化合物1a) を原料として化学的に1位をリン酸化することで、4種のデオキシヌクレオシドの共通原料となるdRPを合成する方法を検討した。
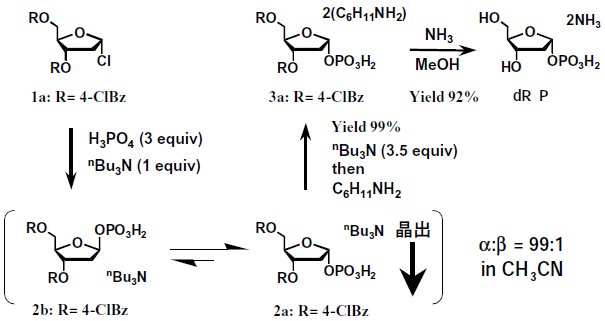
図4 デオキシリボース-1-リン酸の合成法 (異性化晶出法)
ヌクレオシドホスホリラーゼの基質となるのはα体 (図4化合物dRP) のみであり、α体 (図4化合物2aおよび3a) を選択的に合成することが必要とされた。化合物1を塩基存在下にリン酸と反応したところ、縮合反応は進行するもののアノマー位の立体選択性はみられなかった (アノマー比2a:2b=1:1)。
ところが、トリブチルアミン (nBu3N) を塩基としてアセトニトリル中で反応を行ったところ、α体が平衡下に優先して晶析する現象を見出した。この現象を利用して、β体が異性化しながらα体が晶出し、α体を高立体選択的に得ることができた (図4)。
このような反応法は異性化晶出法 (crystallization-induced asymmetric transformation) と呼ばれるが、グリコシル結合の異性化反応やリン酸化糖の合成反応に応用した例としては初めてである6)。
3.デオキシアデノシン (dA)、デオキシグアノシン (dG) 合成反応の開発
大腸菌由来のプリンヌクレオシドホスホリラーゼ (PNPase: EC 2.4.2.1) を利用して、dRPとプリン塩基から、デオキシヌクレオシドを合成する反応は古くから知られていた。しかし、平衡反応であるため、高収率でデオキシヌクレオシドを得るためには、反応条件を工夫する必要があった。特に、水に難溶のグアニンを基質とするdG合成反応では反応収率が上がらなかった。
平衡反応を合成系に傾けるために、合成反応に伴い発生するリン酸を不溶性金属塩として反応系外に排除する方法を検討した。当初、塩化カルシウムなどの水溶性2価金属塩を検討した。塩化カルシウムの添加により、dAの反応収率は80%から96%に向上した。しかし、dGの反応収率は51%から80%と満足できるものではなかった。反応液は、合成反応の進行に伴い晶出したリン酸カルシウムにより粘調なスラリーとなる。水に難溶のグアニンの場合、混合性や溶解性が悪化し、反応が十分進行しなくなると考えられた。また、リン酸カルシウムの濾過分離が困難なことなども、実生産では大きな問題となることが予想された。
そこで、リン酸を反応系外に排除する成分を広く探索したところ、意外なことに水に難溶ということで検討から除外していた水酸化マグネシウムが、効率的に反応を進行させることがわかった。
水酸化マグネシウムは反応開始時に、発生するリン酸見合いの量を一括に添加することができる。水酸化マグネシウムはほとんど溶解していないが、ヌクレオシド合成反応で発生したリン酸はマグネシウム塩として晶出し、反応系外に排除された。
その結果、dAおよびdGを90~96%の反応収率で得ることができた。また、大腸菌由来のチミジンホスホリラーゼ (TPase: EC 2.4.2.4) を用いたチミジン合成反応においても90%の反応収率を得ることができた (図5)。懸念していた濾過性など操作性の問題もなく、実用的な生産方法となった。
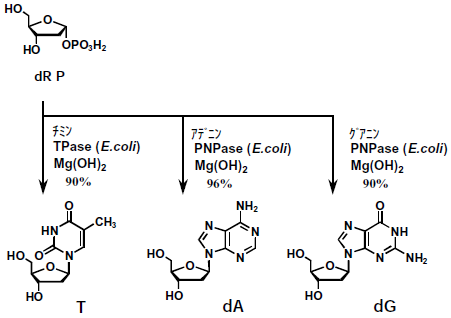
図5 デオKキシヌクレオシド合成反 (T, dA, dG)
4.デオキシシチジン (dC) 合成反応の発見
ヌクレオシドホスホリラーゼには、プリン (アデノシン、グアノシン、イノシン) に働くプリンヌクレオシドホスホリラーゼ (PNPase)、ピリミジン (チミジン、ウリジン) に働くピリミジンヌクレオシドホスホリラーゼ (EC 2.4.2.2)、チミジンに働くチミジンホスホリラーゼ (TPase)、およびウリジンに働くウリジンホスホリラーゼ (UPase: EC 2.4.2.3) が知られているが、シチジンに作用するものは知られていなかった7)。木村らは、プリンおよびピリミジンに働く新規なヌクレオシドホスホリラーゼを報告しているが、この酵素もシチジンには作用しないとされていた8)。
そこでdRPとシトシンからdCを合成するヌクレオシドホスホリラーゼを探索した。しかしながら目的とする活性を持ったヌクレオシドホスホリラーゼはなかなか見出せずにいた。
あるとき、dAやdGの合成検討に用いていた大腸菌由来PNPaseを高発現させた大腸菌をスクリーニングにかけてみたところ、極微量ながらdCが生成することを見出した (図6)。再現性を確認すると、なぜか冷蔵庫で長期保管した菌体を触媒としたときだけdCが生成することがわかった。フレッシュな菌を用いた場合は、生成物としてウラシルやデオキシウリジン (dU) だけが検出された。
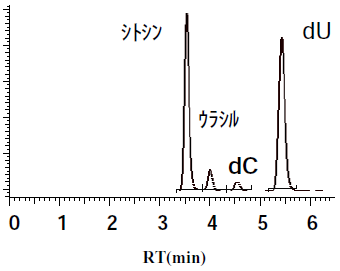

ウラシルやdUが生成する現象は、他の微生物をスクリーニングしたときにも見られていた。これは、夾雑するデアミナーゼ類により、シトシンがウラシルに変換され、さらにウラシルとdRPがUPaseによりdUに変換されるからだと解釈していた (図7ルートa)。大腸菌由来PNPaseの高発現株の結果から、シトシンとdRPがPNPaseによりdCに変換され、dCがウリジンデアミナーゼによってdUに変換されるルートがあるのではないかと思いついた (図7ルートb)。
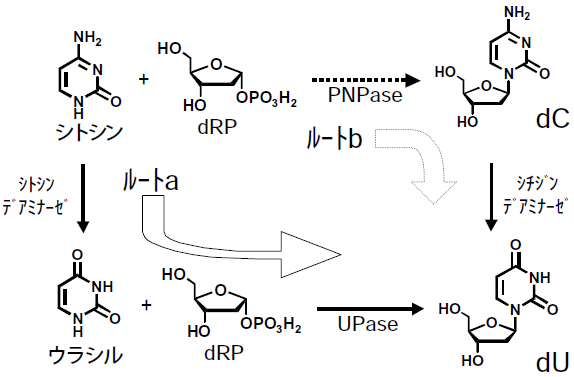
図7 dC合成反応におけるdUの生成ルート
そこで、精製した大腸菌由来PNPaseを用いて反応を行ったところ、dCが単一生成物として得られ、PNPaseにdC合成活性があることが判明した (図8)。菌体触媒として利用するため、シチジンデアミナーゼとシトシンデアミナーゼは、遺伝子破壊法により宿主大腸菌株から排除した。シチジンデアミナーゼとシトシンデアミナーゼを欠損した大腸菌宿主に大腸菌由来PNPaseを高発現させた菌体触媒を用い、dAやdGに準じたdC合成反応を行ったところウラシルやdUはまったく生成せず、反応収率67%でdCを得ることができた (図9)。
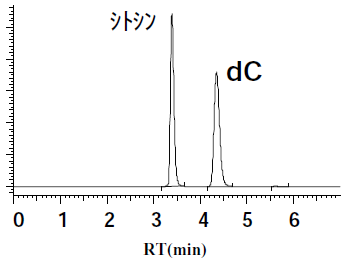

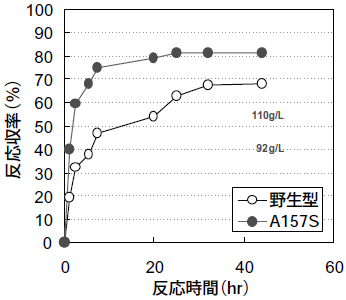
図9 野生型酵素と変異酵素 (A157S) でのdC合成反応
5.進化工学的手法によるdC合成活性の改良
幸運にもdC合成反応を発見することができたが、dAやdGの合成反応と比べると満足のいくものではなかった。dAやdGに比べ合成活性が低いため、相対的に宿主由来のホスファターゼ活性が高くなり、dRPの分解が進んでしまうことが原因であった。そこで進化工学的な手法を用い、dC合成活性の向上を試みた。
エラープローンPCRによりPNPaseにランダム変異を導入し、dC合成活性が向上した変異酵素を選抜した。活性が向上した変異酵素が複数得られたが、単独のアミノ酸置換で最も効果があったのは157位のアラニンをセリンに置換した場合であった。改良したPNPase (A157S) を高発現させた大腸菌を用いて、dC合成反応を行なったところ、活性は約2倍に向上し反応収率は80%となった (図9)。
157位をセリン以外のアミノ酸に置換したところ、セリンのみで顕著な活性向上が見られ、バリンでは活性が大きく低下することがわかった (表1)。また、Lineweaver-Burk プロットからシトシンに対するKm値は、天然型で251 mM、A157S変異酵素で184 mMとわずかに小さくなっていた (図10)。また、dAやdGの合成活性も同時に向上していることがわかった。シトシンアナログには一様に活性が見られたが、5位に置換基が入ったものは活性が低かった。ウリジンやチミジンに対しては殆んど活性が見られなかった (表2)。
表1 変異体のdC合成活性

表2 各種塩基を用いたヌクレオシド合成活性の比較
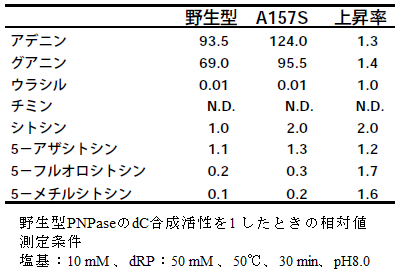
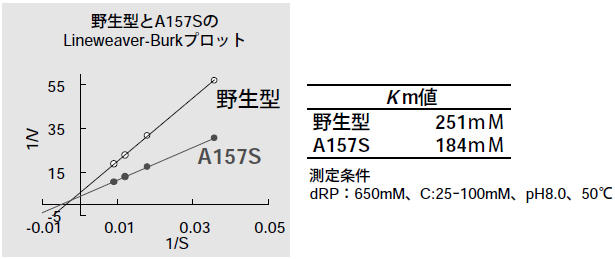
図10 反応速度解析
これら知見と、PNPaseの立体構造データ9)およびすでに提案されているPNPaseの反応機構を10)もとに157位の置換効果を解析した。計算から求めたPNPaseとdCの結合モデルと157位の位置関係を図11 に示した。PNPaseの反応では、205位のアスパラギン酸とプリン塩基の7位の窒素との水素結合が重要とされている。205位のアスパラギン酸は、シトシンの反応においても4位のアミノ基と水素結合を形成すると考えられる。ウラシルやチミンは4位がカルボニル基であることから水素結合を形成できず、反応性が低いと考えられる。157位のアラニンがセリンに置換されると、シトシンの2位のカルボニルとの距離が3.5Å程度になると計算され、水素結合が形成され得る距離になる。この相互作用により、シトシンが反応性の高まる配置に誘導されると解釈すると、157位がバリンに置換されると活性が低下することに納得がいく (図12)。
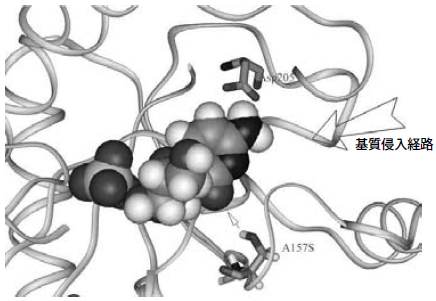
図11 改変酵素の構造
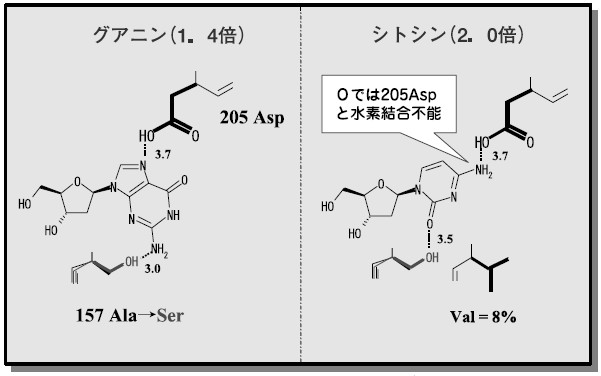
図12 A157Sの活性上昇メカニズム
( )内の数字は野生型に対する活性上昇率
dC合成用の変異型PNPaseは、157位以外の変異を組み合わせることで、現在ではdAやdG並みの反応収率でdCを合成できるようになっている。157位を含め、これら変異により起こる酵素の構造変化と基質特異性の変化については、まだ十分な解析ができていない。さらに解析を進め、PNPaseの構造と基質特異性の関係を解明することで、様々な非天然型ヌクレオシドを合成する触媒の開発が可能になると考えている。
6.おわりに
本稿では、デオキシリボース-1-リン酸 (dRP) の新規な化学合成法とマグネシウムを用いた効率的なヌクレオシド合成法、更にdRPとシトシンからdCを合成する新たな反応を見出したことにより、4種のデオキシヌクレオシドが共通のプロセスで合成できるChemo-Enzymaticな製法の開発について紹介した。
本稿で紹介した製法により、すでにマルチトンスケールでデオキシヌクレオシドが製造販売されている。他のデオキシヌクレオシドが本質的に混入しない製法としてユーザーからの高い評価も得ている。
振り返ってみると製法開発の折々で、非常に幸運な発見が得られたことがポイントであったと感じる。はたしてこれらは幸運だけのなせるワザであったのだろうか。本稿の執筆に際して考え直してみると、それだけではなかったことに気づかされた。既知の情報や思いこみを排除し、地道な実験を繰り返して得られたわずかな手がかりを、形にする努力がそこにはあった。あらためて実験台の前に立ち、試行錯誤を繰り返すことの重要さを再確認する思いである。
謝辞
本研究は、三井化学 (株) 触媒科学研究所、生産技術研究所、マテリアルサイエンス研究所の成果である。これら研究遂行に参加された研究者の方々に感謝をいたします。
文献
1) Nagler, P., Grayson, I.: CHEMISTRY TODAY, Dec., (2003).
2) Sanghvi, Y.: IBC’s 2nd Annual Meeting on Oligonucleotide Technologies, San Diego, CA, Oct., 26 (1998).
3) Krenitsky, T. A., Koszalka, G. W., Tuttle, J. V.: Biochemistry, 20, 3615 (1981).
4) Danzin, C., Cardinaud, R.: Eur. J. Biochem., 62, 365 (1976).
5) (a) Komatsu, H., Awano, H., Tanikawa, H., Itou, K., Ikeda, I.: Nucleosides, Nucleotides & Nucleic Acids, 20, 1291 (2001). (b) Komatsu, H., Awano, H., Ishibashi, H., Oikawa, T., Ikeda, I., Araki, T.: Nucl. Acids Res. Suppl., No.3, 101 (2003).
6) Komatsu, H., Awano, H.: J. Org.Chem., 67, 5419 (2002).
7) (a) 酵素ハンドブック, 朝倉書店 (1982). (b) Robertson, B. C., Hoffee, P. A.: J. Biol. Chem., 248, 2040 (1973). (c) Hoffee, P. A., Blank, J.: Methods in Enzymology, Vol.LI, 437 (1978).
8) Ling, F., Inoue, Y., Kimura, A.: Appl. Environ. Microbiol., 56, 3830 (1990).
9) Mao, C., Cook, W. J., Zhou, M., Koszalka, G. W., Krenitsky, T. A., Ealick, S. E.: Structure, 5, 1373 (1997).
10) Koellner, G., Bzowska, A., Wielgus-Kutrowska, B., Luic, M., Steiner, T., Saenger, W., Stepinski, J.: J. Mol. Biol., 315, 351 (2002).
![]()