【トピックス】
蛍光色素・グアニン塩基間の相互作用に由来する蛍光消光現象を利用した新規遺伝子解析技術
蔵田信也
環境エンジニアリング・産総研分室
1.研究の背景
1.1 オリゴDNAプローブを用いた特定遺伝子の検出
オリゴDNAプローブは、分子生物学の領域において、標的DNAあるいはRNAを特異的に検出する目的で広く用いられてきた。DNAプローブを用いた遺伝子検出手法である各種のハイブリダイゼーション法は、通常、(1) 標的遺伝子を含む核酸混合物の固体表面への固定化、(2) 固定化された核酸とDNAプローブとのハイブリダイゼーション、(3) 未反応DNAプローブの洗浄、(4) ハイブリダイズしたDNAプローブの検出という4つの工程からなり、一般的に煩雑かつ時間のかかる手法である。このため、標的遺伝子に結合することで、プローブの発するシグナルが変化するという性質を有する均一溶液系DNAプローブの研究が盛んに行われている1-4)。均一溶液系DNAプローブを用いたハイブリダイゼーション法では、標的遺伝子を含む核酸溶液とプローブ溶液とを混合し、シグナル変化をモニタリングするだけで、標的遺伝子の検出が可能となるため、一般的なハイブリダイゼーション法で、必要不可欠な固定化・洗浄行程が不要となり、標的遺伝子の検出及び定量を、非常に迅速且つ短時間に行うことが可能となる。
1.2 既存の均一溶液系DNAプローブについて
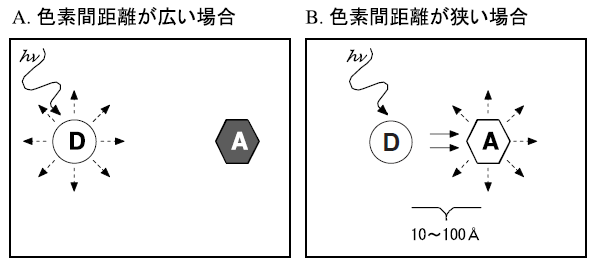
これまで報告されている既存の均一溶液系DNAプローブの多くは、FRET (fluorescent resonance energy transfer5) ) と呼ばれる2種の蛍光色素間の相互作用を利用している。FRET現象は、2種色素が著しく接近した際に、一方の色素 (ドナー) から他方の色素 (アクセプター) に励起エネルギーが転移する現象であり、本現象によりドナー色素の発する蛍光が減少し、アクセプター色素が蛍光を発するようになる (図1)。このFRET現象は、(1) 2つの色素間の距離が10~100 Å (色素ペアによって最適な距離は異なる)、(2) ドナーの蛍光波長と、アクセプターの吸収波長がオーバラップしている場合に観察される。また、FRET現象の効率は、色素間距離の6乗の逆数に比例するため5)、FRET現象を利用した既存の均一溶液系DNAプローブを設計する際、FRETが十分起こるような位置に、2種の色素を正確に配置する必要性がある。このため既存の均一溶液系DNAプローブには、(1) プローブ設計にトライ&エラーが必要、(2) 高コスト (色素が2種必要なこと+プローブ作成にトライ&エラーが必要なため) といった問題点があった。そこで我々は、上記の課題を解決可能な新しい均一溶液系DNAプローブの開発を試みることとした。
![]()
2.新しい均一溶液系DNAプローブ (QProbeTM) について
2.1 蛍光色素とグアニン塩基間の相互作用による蛍光消光現象
新規な均一溶液系DNAプローブの研究開発を行う過程で、オリゴDNAの片方の末端に、ある種の蛍光色素 (BODIPY FL) で標識したDNAプローブを含む溶液と、そのDNAプローブと相補的なオリゴDNAを含む溶液とを混合した際、著しくその蛍光が減少する現象を見出した。この原因を特定するため、各種の検討を行った結果、(1) 本現象は、BODIPY FL とグアニン (G) 塩基間の相互作用に由来する。(2) 蛍光標識した末端塩基の相補的な位置にGが存在したときに最も著しく蛍光消光する、といったことが明らかとなった (図2)6)。これらの知見は、オリゴDNAの末端をシトシン (C) とし、そのC末端に蛍光色素 (BODIPY FL) 標識したオリゴDNAプローブを用い、蛍光の消光を検出することで、標的核酸の特異的検出・定量が可能であることを示唆している。
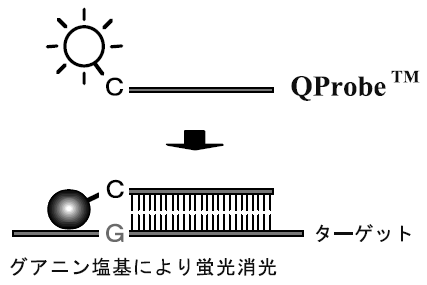
図2 QProbeTMによる遺伝子検出
2.2 QProbeTMの特徴
上記のDNAプローブ (QProbeTM: Quenching Probe) は、C末端が蛍光標識されているのみであり、非常にシンプルな構造を有している。本構造のプローブは、標的遺伝子と結合することで、蛍光色素とG間の相互作用が確実に起こるため、蛍光消光が例外なく発生する。このためQProbeTMは、(1) プローブ設計にトライ&エラーが不要、(2) プローブコストが安価 (プローブの再合成が不要+プローブが安価)、といった特長を有している。また、これまで蛍光消光が顕著な色素は、4種類確認されており、同一反応溶液中に存在する4種の異なる遺伝子を同時に検出することが原理的に可能である。
しかしながら、QProbeTM を用いた手法では、蛍光の減少を検出するため、添加したプローブ量に対して、標的遺伝子量の割合が著しく低い場合 (約1%以下) は、標的遺伝子を検出することが困難となる。この点を考慮し、我々は、QProbeTMと遺伝子増幅法 (主にPCR法) を組み合わせた遺伝子解析技術について検討を進めてきた。PCR法では、非常に高濃度の産物が、最終的にほぼ一定量合成されるため、QProbeTMで精度良く検出することができる。
3.QProbeTM のリアルタイム定量的PCR法への応用
3.1 リアルタイム定量的PCR法について
遺伝子定量技術は、標的遺伝子の存在量や発現量を定量的に把握する目的で、主に研究分野を中心に盛んに用いられている。中でもリアルタイム定量的PCR法は、これまでの遺伝子定量技術と比較して、迅速かつ簡易であり、遺伝子定量を実施する上で切り札的な手法となっている1,4,7-9)。本手法では、遺伝子が増幅する様子をリアルタイムモニタリングすることが必須であるため、遺伝子増幅産物を分離することなく、PCR反応中にモニタリングすることが可能な均一溶液系DNAプローブが、広く汎用されている1,4,9)。
我々は、QProbeTMを応用した新たなリアルタイム定量的PCR法の開発を試み、2つの新規法を開発した。以下、その概要について紹介する。
3.2 QProbe-PCR法
(1) 原理
QProbe-PCR法は、増幅産物の内部配列を認識するQProbeTMを用いて、増幅産物をリアルタイムモニタリングする方法である (図3参照)。このため、本手法では、PCR法において高頻度に増幅される非特異的産物は検出されない。なお、PCRに利用可能な耐熱性DNA合成酵素は、複数種市販されているが、5’-3’ exonuclease活性をもつ酵素は、増幅産物に結合したQProbeTMを分解してしまうため、本法では5’-3’ exonuclease 活性を持たない酵素を使用する必要性がある。
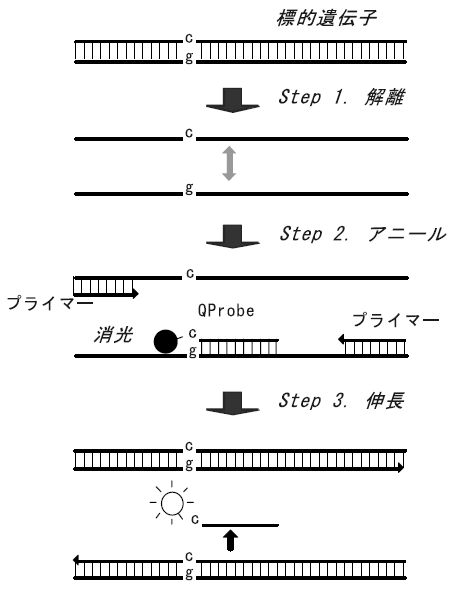
図3 QProbe-PCR法の概要
(2) 分析精度について
QProbe-PCR法の分析精度を客観的に評価するため、標的遺伝子を既知濃度含むサンプルについて分析を実施した。
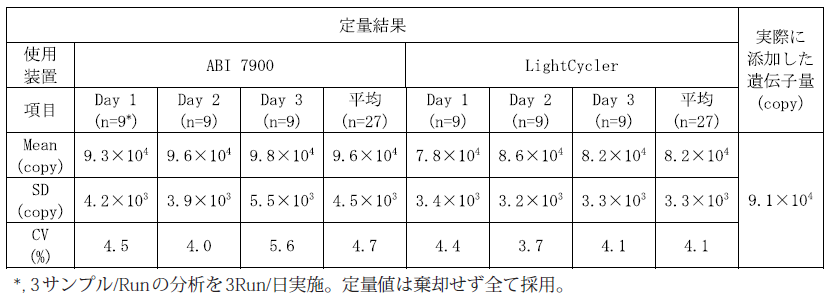
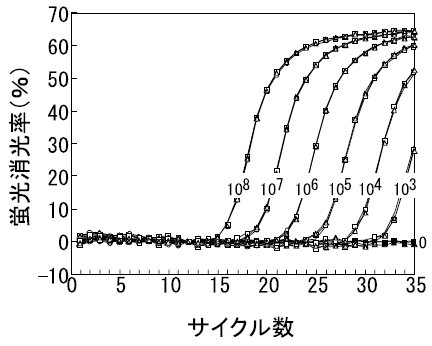
表1に、定量結果を示す。この表から、得られた定量値は、実際に添加した遺伝子量と近い値を示しており、定量誤差を示すCV (%) も4%付近と小さく、定量値の正確性、定量誤差ともに良好であったことが分かる。また、本検討では、2種類のリアルタイム定量的PCR装置を用いたが、ほぼ同様の結果が得られた。図4に、QProbe-PCR法におけるリアルタイムモニタリング結果を示した。この図から、遺伝子増幅を示す曲線は、同一希釈系列 (各3サンプル) では、プラトーに達するまでほぼ重なっていることが分かる。このようにQProbe-PCR法では、非常に精度の高い増幅曲線を得ることができるため、スレッシュホールド値に達するサイクル数にばらつきが生じにくく、結果として、定量誤差が小さく、正確な定量値が得られたものと考えられる。
表1 QProbe-PCR法による定量結果

(3) 特徴
その他のメリットとして、(1) QProbeTMの設計が簡便・確実、(2) 合成したQProbeTMは確実に機能するため、実験系の確立が、簡便、迅速且つ低コストに行える、(3) 特異的産物のみを検出するため特異性が高く、低コピーまで定量可能といった点を挙げることができる。また、本法は、増幅領域が長い場合でも適用可能であることを確認しており (約900 bpまで確認)、本特徴は、特異的なプライマーが、短い増幅領域で設計できない場合、有効であると考えられる。更に、QProbeTMは、相補鎖とハイブリダイズすることで、蛍光シグナルが変化するため、プローブ単独でその機能を確認することができる。このため、系が上手く機能しない場合、その原因をスムーズに特定することができる。
一方、デメリットとして、QProbe-PCR法は蛍光消光をモニタリングする手法となるため、データ解析が特殊となり、リアルタイム定量的PCR装置に付属のデータ解析ソフトを用いることができない点が挙げられる。このため、専用のデータ解析用マクロ (対応機種: LightCycler, ABI7700, ABI7900, iCycler) を作成し、これを使用してデータ解析を実施している。
3.3 QPrimer-PCR法
(1) 原理
QPrimer-PCR法は、5’末端のCを蛍光標識したQProbeTM を、一方のプライマーとして用いる (これをQPrimerと呼ぶ)。このQPrimerを用いてPCR反応を行うと、その反応過程で、蛍光標識されたCの相補的な位置にGが合成される。その結果、蛍光色素とGとが相互作用し、蛍光が消光するため、増幅産物をリアルタイムにモニターすることが可能となる (図5参照)。また、蛍光標識したCに相補的なGが合成されるため、設計したプライマーの5’末端がCでない場合は、その5’末端にCを1つ追加したQPrimerを用いることで、本法は達成される。従って、本法はプライマー配列によりその適用範囲を制限されることはない6)。
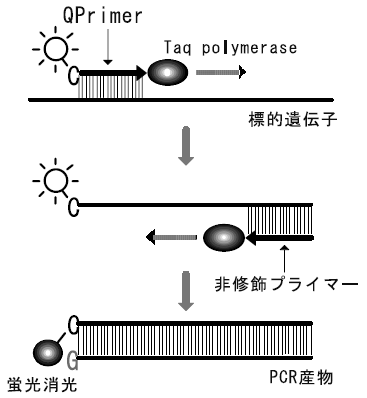
図5 QPrimer-PCR法の概要
(2) 特徴
本法の特徴として、プローブが不要であり、プライマー設計も通常のPCRと同様であるため、実験系の確立がQProbe-PCR法よりも更に、低コスト、且つ簡便に実施できる点を挙げることができる。また、本法においても、QProbe-PCR法と同様、長い増幅産物にも適用可能であることが確認されている (図6)。
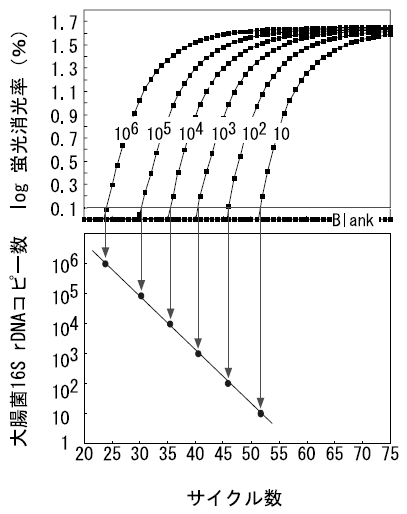

一方、本法のデメリットとして、プライマーから伸長した全ての産物を検出するため、特異的産物だけでなく、非特異的産物を検出してしまう点が挙げられる。しかしながら、本法では増幅産物の解離曲線解析が可能であるため、得られた解離曲線から、非特異的増幅産物の有無を、多くの場合、判別することが出来る。なお、QPrimer-PCR法のデータ解析は、QProbe-PCR法と同様の解析用マクロにて行うことが可能である。
(3) 増幅産物の解離曲線解析について
蛍光色素・グアニン塩基間の蛍光消光現象は、完全に可逆な反応であるため10)、増幅産物が解離することにより、再び蛍光を発するようになる。このため、PCR反応後、温度を変化させながら、蛍光をモニタリングすることで、増幅産物の解離曲線を簡便に得ることができる。PCR反応において増幅される非特異的産物は、プライマー・ダイマーである場合が多く、その長さは、特異的産物よりも通常短い。このため、非特異的産物の解離温度 (Tm値) は、特異的産物のTm値より低い値を示すこととなる。従って、解離曲線解析により産物のTm値を確認することで、特異的産物の有無をある程度判定することができる。しかしながら、特異的産物とほぼ同様のTm値を示す非特異的産物が増幅した場合、解離曲線解析からはその判別が不可能となる。
3.4 各手法の適用先について
QProbe-PCR法は、QProbeTMにて増幅産物の内部配列を認識する手法であるため、基本的に特異的な増幅産物しか検出しない。このため、本法は、特異性が高く、定量範囲も広いことから、1種類の特定遺伝子を確実に定量する目的に適した手法といえる。
一方、QPrimer-PCR法では、増幅産物であれば、特異的産物だけでなく非特異的産物も検出してしまうため、特異性、定量範囲ともにQProbe-PCR法に比べて劣る。しかしながら、共通配列領域を認識するユニバーサルなプライマーを用い、同一機能を有する遺伝子ファミリーを一括定量するようなアプリケーションの場合、QProbe-PCR法では共通配列領域が、3箇所 (プライマー結合領域:2箇所、プローブ結合領域:1箇所) 必要なのに対し、QPrimer-PCR法では2箇所 (プライマー結合領域のみ) でよいため、より簡便に実験系を確立することが出来る。従って、QPrimer-PCR法は、ユニバーサルプライマーを用いた遺伝子ファミリーの一括定量する際に適した手法と考えられる。
また、QPrimer-PCR 法における増幅産物は、1分子毎に、1分子の蛍光色素で標識されるため、蛍光消光率と増幅産物量とは常に正比例の関係が成り立つ。このため、本法にて増幅産物量をリアルタイムに定量することが可能である。このユニークな特徴を利用することで、分子生物学的な微生物相解析において問題となるPCRバイアスやアーティファクト生成を回避することが可能であり (データ不掲)、微生物相解析を精度良く実施するためのツールとして非常に有効であると考えられる11)。
4.QProbeTM を用いたSingle nucleotide polymorphisms (SNPs; 一塩基多型) の解析手法
(1) SNPs解析の意義
SNPs (Single nucleotide polymorphisms) とは、個人間の一塩基の違いを意味しており、その違いが全人口中1%以上の頻度で存在する場合、SNPsと呼んでいる12,13)。SNPsの解析は、疾病関連遺伝子の特定、薬剤応答性・副作用の診断、創薬ターゲットの探索などにつながると考えられるため、医療研究分野を中心に急速に浸透しはじめている7,14)。これら基礎的な研究により臨床的に意味のあるSNPsが、今後数多く特定され、そのSNPsを診断することにより、個々の遺伝情報に基づく最適な医療行為 (テイラーメード医療) を行うことが可能になると広く予想されている。以上の背景から、臨床分野においてSNPs解析技術の重要性は、飛躍的に高まってゆくものと考えられる。
(2) QProbeTMを用いたSNPs解析手法の原理
QProbeTMの蛍光消光現象は完全に可逆な反応であるため、QProbeTMと標的遺伝子をハイブリダイズさせた後、徐々に温度を変化させながら、連続的に蛍光をモニタリングすることにより、簡単に解離曲線を作成することができる。QProbeTM を用いたSNPs解析手法は、この解離曲線解析により実施する。その原理を図7に示した。まずSNPs部位を覆うようにQProbeTMを作成する。次に、SNPs部位を含む領域をPCR増幅し、増幅終了後、QProbeTMと増幅産物の解離曲線解析を実施する。解離曲線解析は、PCR増幅前にQProbeTMを添加することにより、非解放で解析することができる。
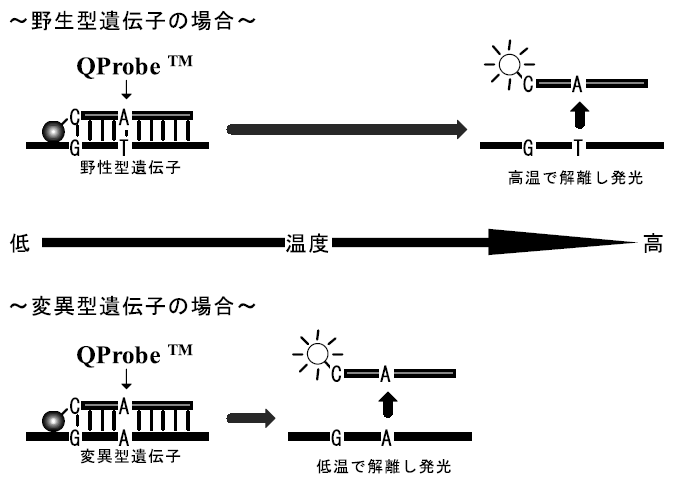
図7 QProbeTMを用いたSNPs解析の概要
QProbeTMは、一方の遺伝子型 (図では野生型遺伝子) と完全相補的となるよう作成するため、他方の遺伝子型についてはQProbeTMとの間には1つのミスマッチ存在することとなる。ミスマッチが存在する場合、2本鎖DNAの熱力学安定性が低下するため、ミスマッチを含む遺伝子型とQProbeTMとの解離は、完全相補的な遺伝子型との解離よりも低い温度で起こる。解離が起これば蛍光が増大するため、どの温度領域でQProbeTMの蛍光が増大したかを確認することで、遺伝子型を特定することができる。
(3) SNPs解析用のQProbeTMの設計
QProbeTMが20塩基前後の場合、1つのミスマッチによって、約5~10℃程度のTm値の差 (ΔTm) が観察される場合が多い。ただ、QProbeTMのどの位置に、ミスマッチを配置するかによって、ΔTmが大きく異なるため、事前にΔTmを予想して、SNPs解析に好適なQProbeTM を設計する必要性がある。この目的のため、我々は、簡便にΔTmを予測することが可能なソフトを独自に開発した。本ソフトを用いることで、SNPs解析を実施可能なQProbeTMを、簡便且つ、高い確率で設計することができる。
(4) QProbeTMを用いたSNPs解析手法の実際
実際の適用例を、図8として示した。ここで用いたQProbeTMは、野生型遺伝子に対して完全相補的であり、変異型遺伝子に対しては1つのミスマッチを含むこととなる。このため、野生型遺伝子よりも、変異型遺伝子のほうが低い温度で解離し、蛍光を発するようになると予想される。上図 (図8A) が解離曲線で、下図 (図8B) が解離ピークを示している (解離ピークは、解離曲線を微分したものであるため、その頂点は、最も蛍光強度変化の著しい温度を示しており、この温度はTm値とほぼ一致する。)。図8Bより、QProbeTMと野生型遺伝子との解離ピークの頂点 (Tm値) は、約60℃付近を示しているのに対し、QProbeTMと変異型遺伝子では約50℃付近のTm値を示している。この結果より、遺伝子型を、Tm値の差から明瞭に特定可能であることが分かる。また、野生型遺伝子と変異型遺伝子をヘテロで含む場合、その解離曲線は、2つの解離曲線を丁度合わせた形状となり、解離ピークが2つ現れる。このように、解離ピークが2つ現れた場合は、2つの遺伝子型が存在することを確定することができる。
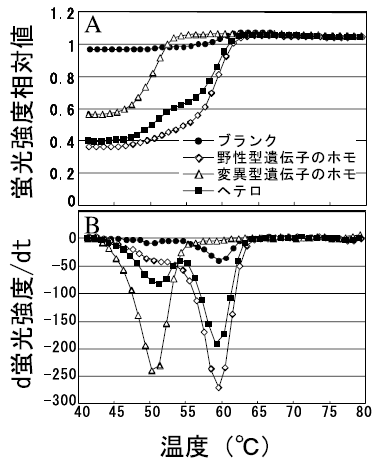
図8 QProbeTMを用いたSNPs解析結果 (使用装置: LightCycler)
(5) QProbeTMを用いたSNPs解析法の適用先
本SNPs解析手法は、SNP毎に異なるQProbeTMを用意する必要性のあることから、1SNP 当たりの解析回数は少ないが、非常に多種類のSNPsを解析するような場合、用意したQProbeTMの多くが未使用となるため、高コストとなる。このため、本法は、網羅的なSNPs解析には、不向きであると考えられる。しかしながら、(1) ゲノムDNAから1ステップで解析できるため、操作が簡易かつ迅速 (増幅法がPCR法の場合、約0.5~2時間で完了)、(2) 1SNPの解析を1種類のQProbeTMで実施できるため低コスト (プローブを使い切る場合のプローブコストは約3~10円/SNP)、(3) 遺伝子増幅に必要な試薬類とQProbeTM以外に、特別な試薬を必要としないため低コスト、(4) 1反応系内で複数SNPsの同時タイピング可能であり、試薬費や鋳型使用量を削減可能、(5) 増幅産物が増えれば、SNPs解析が実施できるため、PCR条件 (特にアニーリング) の許容範囲が広い、といった特長を本法は有しており、決まったSNPを複数回実施するような場合に、好適な手法であると考えられる。また本手法は、再現性が非常に高く、一般的にタイピングが困難なヘテロの場合でも、確実にタイピングすることができる (データ不掲)。以上より、QProbeTMを用いたSNPs解析手法は、コストメリットが厳しく要求され、絶対に間違いが許されない臨床検査の分野において、非常に有効な手法であると認識される。
5.おわりに
21世紀は生命科学の世紀であり、バイオテクノロジーの世紀である。バイオテクノロジーの進歩は、人間生活の基本である『生きる』、『食べる』、『暮らす』の3場面のあり方を抜本的に変えるインパクトを持ちうる極めて大きな技術革新をもたらす (ライフサイエンス・サミット大会宣言抜粋)。
バイオテクノロジーとは、生命の持つ機能を実用に重きをおいて応用する科学技術である。生命は、設計図である遺伝子を例外なく有しており、本生体分子は、遺伝子を設計図として生成されるタンパク質と共に、『生命』が『生命』であるための根源的構成要素である。従って、遺伝子ならびにタンパク質を詳細に追跡・解析することが生命現象を正確に理解することにつながり、その結果はじめて生命機能を高度に利用することが可能となる。以上より、遺伝子の解析技術なくして今日のバイオテクノロジーの発展は考えられず、今後も本解析技術がバイオテクノロジーの基盤を支えてゆくことは確実と思われる。
しかしながら、既存の遺伝子解析技術は、1) 操作が煩雑、2) 時間がかかる、3) イニシャルコスト (装置)、ランニングコスト (試薬) ともに高価、4) 精度が目的に合わない、等の問題点を有しており、これらが障害となって、遺伝子解析技術の導入がなかなか進まないケースが決して少なくない。前述のように遺伝子解析技術は、バイオテクノロジーを基盤から支える必須な技術であるため、上記の問題点が解決され、幅広い産業分野に本基盤技術が活用されるようになれば、バイオテクノロジーを加速度的に進展させることが可能となり、その結果、新規な市場を数多く創出できるものと考えられる。
しかしながら、現在、遺伝子解析が実施されている国内の現場では、解析装置、試薬など、外国製品に依存する場合が非常に多く、それは結果的に、外国企業の競争力を強め、国外の新規市場を開拓していることにつながりかねない。このため、バイオテクノロジーの基盤となる優れた純国産の遺伝子解析技術を開発することが、我が国のバイオテクノロジー分野における国際競争力を向上させるために、重要且つ緊急な課題であると考えられる。
これまで述べてきたように、QProbeTMを用いた遺伝子解析手法は、基本原理が非常にシンプルであるため、簡便、低コストかつ信頼性の高い手法である。今後は、純国産の遺伝子解析技術である本手法を、更に応用発展させ、我が国のバイオテクノロジーの発展に少しでも貢献できるよう努力してゆくつもりである。
本研究は、(独) 産業技術総合研究所と環境エンジニアリング (株) との共同研究による成果である。これらの研究開発に参加された方々、また各方面でご協力いただいた方々に深く感謝いたします。
文献
1) Tyagi, S., Kramer, F. R.: Nature Biotechnol., 14, 303 (1996).
2) Morrison, L. E., Halder, T. C., Stols, L. M.: Anal. Biochem., 183, 231 (1989).
3) Cardullo, R. A., Agrawal, S., Flores, C., Zamecnik, P. C., Wolf, D. E.: Proc. Natl. Acad. Sci. USA, 85, 8790 (1988).
4) Holland, P. M., Abramson, R. D., Watson, R., Gelfand, D. H.: Proc. Natl Acad. Sci. USA, 88, 7276 (1991).
5) Stryer, L., Haugland, R. P.: Proc. Natl. Acad. Sci. USA, 58, 719 (1967).
6) Kurata, S., Kanagawa, T., Yamada, K., Torimura, M., Yokomaku, T., Kamagata, Y., Kurane, R.: Nucl. Acids Res., 29, E34. (2001).
7) Mackay, I. M., Arden, K. E., Nitsche, A.: Nucl. Acids Res, 30, 1292 (2002).
8) Ishiguro, T., Saitoh, J., Yawata, H., Yamagishi, H., Iwasaki, S., Mitoma, Y.: Anal. Biochem., 229, 207 (1995).
9) Wittwer, C. T., Herrmann, M. G., Moss, A. A., Rasmussen, R. P.: Biotechniques, 22, 130 (1997).
10) Torimura, M., Kurata, S., Yamada, K., Yokomaku, T., Kamagata, Y., Kanagawa, T., Kurane, R.: Anal. Sci., 17, 155 (2001).
11) Kanagawa, T.: J. Biosci. Bioeng., 96, 317 (2003).
12) Lai, E., Riley, J., Purvis, I., Roses, A.: Genomics, 54, 31 (1998).
13) Sachidanandam, R., Weissman, D., Schmidt, S. C., Kakol, J. M., Stein, L. D., Marth, G., Sherry, S., Mullikin, J. C., Mortimore, B. J., Willey, D. L.: Nature, 409, 928 (2001).
14) Horikawa, Y., Oda, N., Cox, N. J., Li, X., Orho-Melander, M., Hara, M., Hinokio, Y., Lindner, T. H., Mashima, H., Schwarz, P. E.: Nature Genet., 26, 163 (2000).
![]()