【トピックス】
脊椎動物におけるDNAのメチル化修飾とその制御
田嶋正二、末武 勲、木村博信
阪大・蛋白質研究所
1.はじめに
原核生物におけるシトシン塩基のメチル化修飾酵素は一般に、同じ塩基配列を切断する制限酵素とともに発現していて、自己のDNAを制限酵素による切断から保護している (restrictionmodification系)。これは、ファージなど外来から侵入するDNAに対する防御機構と考えられている。これに対して、真核生物におけるシトシン塩基のメチル化修飾は転写制御機構の一つとして進化した。ただ、単細胞である出芽酵母ゲノムにはシトシンのメチル化酵素遺伝子は欠落していて、したがって、DNAのメチル化修飾はない。進化がすすんだ高等植物や脊椎動物では、ゲノムの塩基量が、トランスポゾンや繰り返し配列などを取り込むことにより飛躍的に増大したが、シトシンのメチル化修飾はそれと併行して密度と分布域ともに飛躍的に増大している。これは、進化の過程でゲノムに取り込まれたトランスポゾンの転写抑制、繰り返し配列の安定化、さらには、多様化した体制を整然と保つために、細胞や組織で不要な遺伝子群を完璧に抑制する道具として利用されたことによると考えられる。トランスポゾンや繰り返し配列をゲノムに取り込み、塩基量を増大させたことは、種全体にとっては体制を複雑化させることに有利に働いたが、個々の個体にとっては異常な遺伝子の発現の可能性を増したという負の側面をもつ。この負の面をDNAメチル化によるサイレンシング機構が補償していると考えられる。哺乳類の体細胞ゲノムDNAではメチル化可能な配列のうち約80%がメチル化修飾を受けているとされる。
本稿では脊椎動物、特に研究の進んでいる哺乳類のDNAメチル化の制御機構を中心にして解説したい。上述のようにDNAのメチル化は転写制御機構の一つとして、遺伝子のサイレンシングに利用されているが、哺乳類ではさらに、ゲノム・インプリンティング (性特異的なゲノムのメチル化模様の違いにより、対立遺伝子の一方の発現が抑制される現象) や、X染色体の不活性化に寄与している。また、DNAのメチル化は癌化や老化などの病理現象にも深く関わっている。DNAメチル化模様の調節機構を解明するには、DNAメチルトランスフェラーゼがどのように標的のゲノム領域を認識してメチル化し、模様を細胞系列特異的に維持しているのかを明らかにする研究が重要となる。
2.DNAのメチル化修飾
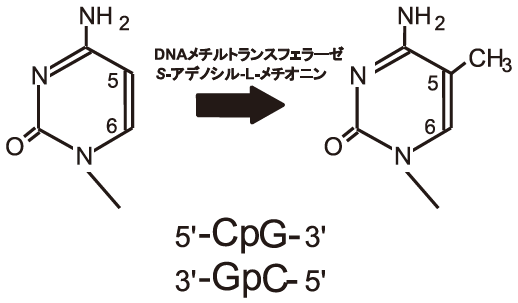
DNA中のシトシン塩基は生理条件下でメチル化修飾を受ける。1948年にHotchkissは仔ウシ胸腺に含まれるDNA塩基を薄相クロマトグラフィーで分析し、A、G、C、T以外にCの修飾塩基が含まれることを見出し、これが5-メチルシトシン (5mC) であると予想した。1951年にはWyattは動物組織や植物組織のゲノムの塩基分析で5mCが含まれていることを証明した。その後、5mCは原核生物である細菌ももっていることが明らかになり、真核生物では菌類から高等植物、哺乳類までほとんどの生物のゲノムに5mC修飾が見つかっている。例外的に、線虫や出芽酵母のゲノムに5mCは存在しない。脊椎動物では、CとGの順に並んだCpG配列があると、DNAメチルトランスフェラーゼはシトシン塩基の5位の炭素にS-アデノシル-L-メチオニンからメチル基を転移、付加する (図1)。

DNAのメチル化修飾は転写を抑制するが、その阻害様式には直接的なものとクロマチン構造の変化を伴う間接的なものの二通りある1)。転写因子結合モチーフかその付近にCpG配列があり、それがメチル化修飾を受けると、一部の例外を除いて、多くの場合転写因子はモチーフへの結合が阻害される。その結果、転写活性が阻害されると考えられるが、生体内でこのメチル化修飾による直接的な阻害機構が働いていることがはっきりしている例は、実は少ない。生体内で結合モチーフのメチル化が直接転写活性に影響する例として、転写因子CTCFがある。CTCFはDNAに結合してエンハンサー効果を遮断するインスレーター機能を持つが、結合モチーフがメチル化されているとDNAに結合できないために、インスレーター効果を発揮できない。ゲノムインプリントを受ける遺伝子であるIgf2とH19の両遺伝子の間の特定の配列 (DMR、differentially methylated region) は雄に特異的なメチル化修飾を受けるが、そこにCTCF結合配列が存在する。ここにインスレーター活性を持つCTCFが結合するか否かにより、H19遺伝子下流に存在するエンハンサーがIgf2とH19のどちらの遺伝子に作用するのかが制御されている2,3)。
一方、間接的な転写抑制機構にはメチル化修飾を受けたCpGを特異的に認識して結合する一群の蛋白質が中心的な役割を担っている。これらのDNA結合蛋白質は共通して、MBD (methyl-CpG-binding domain) と呼ばれるメチル化された配列を特異的に認識する領域を持っている。これらの蛋白質はヒストンを脱アセチル化やメチル化する酵素をメチル化されたDNA領域にリクルートすることによって、結合領域を不活性なヘテロクロマチン構造に変換させ、これにより遺伝子の発現を抑制している1)。
3.DNAメチル化模様の形成
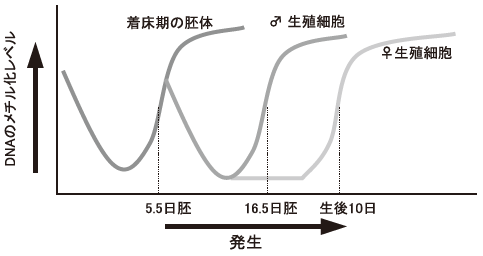
マウスなど哺乳類のゲノム全体のメチル化レベルは、組織が分化してしまうと比較的一定であるが、胚発生、生殖細胞の成熟過程で大きく変動する4)(図2)。これに対して、アフリカツメガエルやゼブラフィッシュなどの非哺乳類の脊椎動物では、限定された範囲の遺伝子領域では、細胞の分化にともなってDNAのメチル化状態は変化するものの、初期発生の過程でゲノム全体のメチル化レベルが大きく変化することはない。

3-1 DNAメチル化模様を形成するDNAメチルトランスフェラーゼ
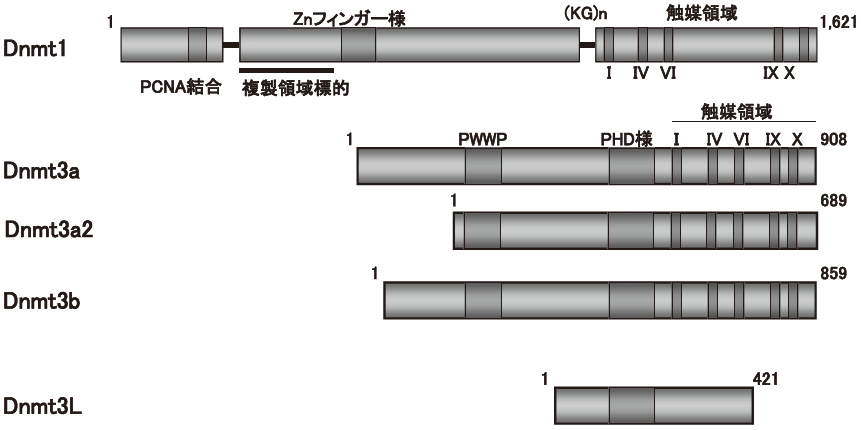
ゲノムのメチル化模様を形成する (de novo型) DNAメチルトランスフェラーゼにはDnmt3aとDnmt3bの二つがある (図3)。両酵素はともに約100 kDaの蛋白質で、別々の遺伝子にコードされている。いずれもC末端半分に触媒活性責任領域がある。N末端側半分には、両酵素に共通してCys残基に富むPHD (plant homeodomain) とPWWP領域が存在する。PHDは多くのクロマチン結合蛋白質に見られる配列で、他の蛋白質との相互作用に寄与している。また、Dnmt3bのPWWP領域は立体構造が解かれていて、新たなDNA結合モチーフであることが報告されている5)。両酵素が生体内でde novo型のDNAメチル化活性を示すことは、遺伝子をノックアウトしたマウスで示された。両酵素とも欠く胚は着床期のメチル化模様の書き込みが障害され、着床後間もなく致死となる。Dnmt3a遺伝子だけをノックアウトすると、生まれるが生後4週で痩せ衰えて死んでしまい、Dnmt3b遺伝子のノックアウト・マウスでは生まれる直前に致死となる6)。このことから、Dnmt3aとDnmt3bはお互いに機能を部分的に相補していることがわかる。Dnmt3bは選択的にセントロメア領域のマイナー・サテライトをメチル化する。ICF (immunodeficiency, centromeric region instability, and facial anomalies) 症候群と呼ばれる遺伝病は1、9、16染色体のセントロメア付近のヘテロクロマチン領域が低メチル化状態になることにより発症するが、これはDnmt3b遺伝子の変異が原因である。着床期の胚体ではDnmt3bが特異的に高発現しているのに対して、Dnmt3aの発現はDnmt3bの発現が低下した10.5 日胚以降となる7)。両酵素の発現時期の違いがメチル化する領域の違いに寄与していると考えられる。組換体を用いたDNAメチル化活性測定から、in vitroにおいても両酵素はde novo型の活性を示すことが明らかとなっている8)。

哺乳類ゲノムのメチル化模様は、インプリント遺伝子を含めて生殖細胞で一旦消去され、新たに書き込まれる。生殖細胞におけるゲノムのメチル化模様の書き込みにはDnmt3aの遺伝子産物が主役であることがノックアウト・マウスの実験により示されている9)。しかし、精原細胞では全長のDnmt3aはほとんどまったく発現せず、N末端の219アミノ酸残基を欠いたDnmt3a2と呼ばれるアイソフォームが高発現している10,11)。この時期のゲノム全体のメチル化に寄与しているのはDnmt3aではなく、Dnmt3a2である。生殖細胞でのDNAメチル化模様の形成にはDnmt3L (Dnmt3-like) と呼ばれる、Dnmt3aやDnmt3bと構造的に似てはいるがDNAメチル化活性を持たない因子を要求する12,13)。Dnmt3Lはin vitro でDnmt3a、Dnmt3a2、Dnmt3bのDNAメチル化活性を促進することが報告されている11,14,15)。雄性生殖細胞でDnmt3Lを絶対的に要求するのは、N末端を欠くDnmt3a2がDnmt3aとは違って、生理的な塩環境下ではDNAメチル化活性を発揮できないが、Dnmt3Lの存在下では有意な活性を示すことによる11,15)。
Dnmt1は後述のように、一旦形成されたメチル化模様を維持する型のDNAメチルトランスフェラーゼと考えられているが、最近ゲノムにメチル化模様を新規に書き込む可能性が指摘されている16)。これまでもin vitroでは新たにメチル化模様を書き込むde novo型の活性を持つことが報告されているので、そのde novo型の活性が生体内でも発揮されていることは充分あり得る。
3-2 Dnmt3aとDnmt3bのDNAメチル化活性の異同
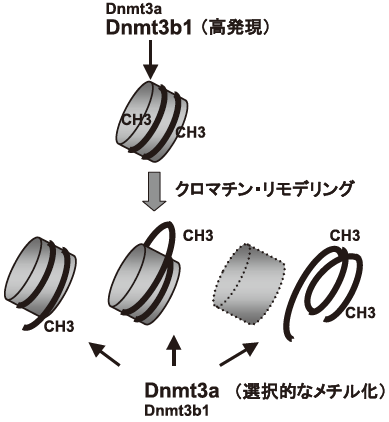
Dnmt3aとDnmt3bは、生体内では一部ゲノムの異なる領域をメチル化しているが、in vitro で裸の2本鎖DNAを基質とした場合はお互いによく似たDNAメチル化活性を示す8,17)。しかし、生体内でDNAが組み込まれているヌクレオソームをメチル化受容基質として両酵素の活性を比較すると、両者の違いは際立つ。両酵素ともヌクレオソームの形成によって活性が著しく阻害されるが、Dnmt3bがヌクレオソームのコア領域をある程度メチル化できるのに対して、Dnmt3aはその活性が著しく低い。一方、Dnmt3aはヌクレオソーム内でDNAが裸で露出している領域を効率よくメチル化する。Dnmt3bの発現量が時期や細胞特異的に厳密に調節されているのに対して、Dnmt3aは、発現量は低いものの様々な細胞でユビキタスに発現している。発生期での発現のタイミングや、生殖細胞で見られた組織特異的な発現様式の違い、それとヌクレオソームDNAをメチル化する活性の違いが総合されることで、両酵素がゲノムの異なる領域をメチル化していると考えられる18)。Dnmt3bが主に繰り返し配列を含むゲノム・ワイドな領域のメチル化模様の形成に、そして、Dnmt3aが特定領域のDNAメチル化に寄与していると考えたい (図4)。

3-3 クロマチン構造変化とDNAメチル化形成
シロイヌナズナでSWI2/SNF2 型のクロマチン・リモデリング因子をコードするDDM1遺伝子の働きが障害されると、ゲノム全体のメチル化が低下する19)。マウスのDDM1遺伝子ホモログであるLsh 遺伝子をノックアウトすると、やはり、繰り返し配列をはじめゲノム全体のメチル化が低下する。LshはDnmt3aやDnmt3bと協同してゲノムに新たなメチル化模様を書き込んでいる20)。また、ヒトのクロマチン・リモデリング因子であるATRX遺伝子に変異が起きると、リボソームRNA遺伝子では低メチル化が、Y染色体特異的なサテライトDNAやテロメア近傍の繰り返し配列では高メチル化が引き起こされる21)。リンカー・ヒストンと呼ばれるヒストンH1は、ヌクレオソーム間のtransな相互作用やパッケージングに寄与しているが、マウスに6つあるヒストンH1遺伝子のうち3つをノックアウトし、その発現量を半分に減らした胚性幹細胞 (ES細胞、embryonic stem cells) ではクロマチンの構造が大きく変化して、インプリント遺伝子やX染色体上などの限られた遺伝子の発現とメチル化状態が変化していることが報告されている22)。このように、クロマチンの構造を変化させる様々な遺伝的な変異がDNAのメチル化状態に影響を与えることが報告されている。
3-4 ヒストン修飾とDNAメチル化形成
アカパンカビのゲノムのメチル化修飾は、ヒストンH3の9番目のLys残基 (H3K9) をメチル化する酵素をコードするDim-5 遺伝子をノックアウトすると消えてしまう23)。植物ではCpG配列に加え、CpNpG、CpNpN配列中のシトシンもメチル化修飾を受けるが、シロイヌナズナでは、CpNpGをメチル化するDNAメチルトランスフェラーゼ (CMT3) はメチル化されたヒストンH3K9とヒストンH3の27番目のLys残基 (H3K27) に結合し、これによって近傍のDNAをメチル化することが報告されている24)。ポリコーム群 (PcG) 蛋白質と呼ばれる一群の蛋白質複合体は遺伝子の発現抑制に寄与しているが、哺乳類のポリコーム群蛋白質複合体は、構成因子であるEZH2がヒストンH3K27をメチル化し、付近の領域をヘテロクロマチン化してサイレンシングを引き起こす。このEZH2は、Dnmt1、Dnmt3a、Dnmt3bをリクルートするプラットフォームにもなっていて、結合領域付近のDNAメチル化修飾に寄与している25)。マウスではヒストンH3K9をトリメチル化する酵素をコードするSuv39h1とSuv39h2遺伝子を変異させると、ペリセントロメア領域のメジャー・サテライト領域のメチル化が低下することが報告されている26)。このように、ヒストンのメチル化修飾、特にヒストンH3のK9とK27のメチル化修飾とゲノムのメチル化には強い因果関係が認められるが、その基盤となる分子機構については今後の課題として残されている。
3-5 特異的な配列のDNAメチル化形成
これまで述べたメチル化模様の創生は、クロマチンやヒストン修飾などの変化に応じた、おそらく比較的広い領域のDNAメチル化模様の書き込み調節である。では、特定の遺伝子を標的にしたメチル化模様の書き込み機構が存在するのだろうか。組換Dnmt3aは、標的のCpGに加えて周り数ヌクレオチドの範囲も認識してメチル化するとの報告もあるが、標的配列が短いことや、Dnmt3aとDnmt3bの両者を比較しているわけではないので、この性質だけでメチル化する標的配列の特異性を論ずることは難しい。
癌遺伝子であり転写因子でもあるc-MycはDnmt3aのCys残基に富むPHD (plant homeodomain) 様配列に結合して、Dnmt3aをp21Cip1遺伝子のプロモーター領域にリクルートし、付近のメチル化を誘導して、p21Cip1遺伝子の発現を抑制することが報告されている27)。また、維持型のメチル化酵素であると考えられているDnmt1がp53の標的配列にリクルートされて、その付近をメチル化することが報告されている16)。クロマチン・リモデリング複合体NoRCはリボソームRNA遺伝子の転写抑制に寄与するが、その構成因子Tip5はDnmt1とDnmt3bをリクルートして遺伝子のサイレンシングに貢献しているなど28)、DNA配列特異的に結合する因子がDNAメチルトランスフェラーゼをリクルートして、メチル化する領域を規定する機構が報告されてきている。同様の機構が今後さらに報告され、DNAメチル化模様の調節に重要性を増すものと予想される (図5)。
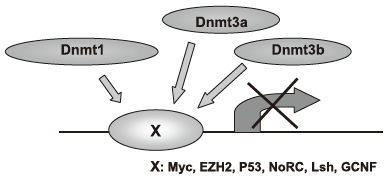

植物には、短い2本鎖RNAが相補する特定の塩基配列をメチル化する機構 (RNAi; RNA interferences) がある。この短いRNAがどのように標的の塩基配列をターゲットするのか良くわかっていないが、クロマチン・リモデリング因子の一つDRD1や植物に特異的なRNAポリメラーゼIVなどが介在していることが報告されている29)。同じように、動物細胞でもRNAiがゲノムのメチル化配列の標的に寄与するのかについてはまだ確定していない。
4.ゲノムメチル化模様の維持
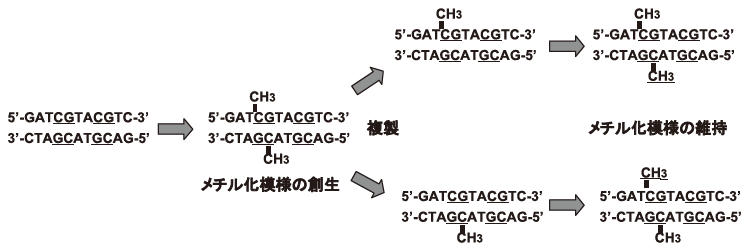
DNAのメチル化修飾がエピジェネティックスの定義を満たすのは、その模様が次世代の細胞に伝えられる機構があることによる。ゲノムに形成されたメチル化模様が次世代の細胞に維持されるのは、最初にcDNAが単離されたDNAメチルトランスフェラーゼDnmt1の働きによる。Dnmt1はDNA複製やDNA修復直後に生じる2本鎖DNAに存在する、片方がメチル化されたCpG対 (ヘミメチル化DNA、図6) を選択的にメチル化して、DNAメチル化模様を細胞系列特異的に維持している。Dnmt1遺伝子のノックアウト・マウスでは、着床後にゲノム全体に起きるメチル化修飾の時期にメチル化インプリントの消失や不活性X染色体の再活性化が引き起こされ、胎生9.5日で致死となる。

4-1 Dnmt1分子の機能構造領域
マウスDnmt1は1,621アミノ酸残基からなる大きな分子で、分子内を、機能的、エキソン配置、構造的な基準から3つの部分に分けることができる (図3)。
N末端約290 アミノ酸残基からなる配列には、複製に必須なPCNA (proliferating cell nuclear antigen)30)、転写抑制因子DMAP1 (Dnmt1 associated protein)31)、DNAに結合する配列がある32)。PCNAは複製フォークに結合してDNAポリメラーゼやDnmt1などを結合するプラットフォームとして機能するが、この配列と重なるようにしてDNA結合配列が存在する。このDNA結合活性はATに富むDNAの狭い溝に結合し、PCNAへの結合と競合する。ATに富む配列内のCpGは多くの場合メチル化修飾を受けていることから、N末端付近に存在するDNA結合配列は、PCNAを要求しない短い塩基除去修復箇所にDnmt1をリクルートするために働いていると推察される32) (図7)。このN末端領域は、ヘミメチル化CpGを認識してメチル化する触媒活性には影響を与ないことを考えると33)、Dnmt1分子の局在を制御し、機能を調節する因子をリクルートするプラットフォームの役割を担っているのであろう。
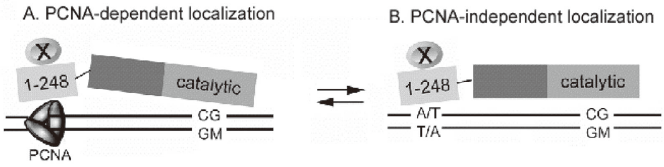

C末端500アミノ酸残基は、細菌のシトシンメチル化酵素が共通してもつモチーフを保存するメチル化触媒活性責任領域である。分子中ほどの領域は、C末端領域とLys-Gly (KG) が繰り返す配列で分けられている。Znフィンガー様の配列をもち、この配列を介してヒストン脱アセチル化酵素HDAC、HP1 (heterochromatin protein1)、MBDの一つMeCP2など、転写抑制に関わる因子と結合している。この中間領域は、ヘミメチル化CpGの認識や複製領域へのリクルートなど、Dnmt1の活性を調節する上で特に重要であるが、この中間領域の大部分を欠くとDnmt1は活性を示さないことから機能解析が進んでいない。おそらくこの中間の領域はC末端の触媒領域とは分子内で相互作用していて、機能的に不可分であると考えられる。
4-2 Dnmt1の酵素的な性質とメチル化模様維持機構
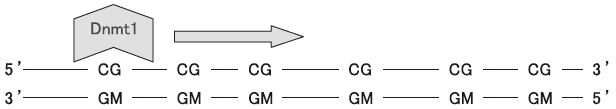
Dnmt1はメチル化模様の維持に中心的な役割をはたす。したがって、Dnmt1がヘミメチル化CpGを特異的に認識する機構の解明が最も重要な問題であるが、これについては現在のところほとんど解明されていない。メチル化模様を維持するためにはもう一つ、複製が終了した娘鎖上のヘミメチル化CpGをDnmt1は順次メチル化する性質が必要となる。Dnmt1はN末端領域でPCNAと結合することによって複製鎖上をDNAポリメラーゼなど複製因子複合体とともにスライドして、合成されたばかりのヘミメチル化CpGを順次メチル化していくモデルが考えられている。しかし、我々の結果では、結合配列を含むN末端領域を欠くDnmt1でも、DNA2本鎖の片一方の鎖をスキャンして、遭遇するヘミメチル化CpGを順番にメチル化する性質を保持していた (図8)33)。したがって、この特性は複製フォーク上でPCNAと結合することに依存したものではなく、酵素自身が持っている性質であるといえる。

一旦形成されたメチル化模様はDNAが複製される過程で正確に次世代の細胞に伝えられている34)。しかし、Dnmt1がヘミメチル化CpGをどの程度の精度でメチル化するのか調べると、どうも20に1回はメチル化を入れ損なっている33,35)。de novo型酵素をコードするDnmt3b遺伝子をノックアウトした細胞では、継代を重ねるとゲノムのメチル化模様が低下していくことが観察されているので36)、次世代にメチル化模様を正確に伝えるためにはDnmt1だけでなく、他のDNAメチルトランスフェラーゼの助けが必要と考えられる。実際、Dnmt1のN末端領域にはDnmt3aとDnmt3bが結合することが報告されている37)。Dnmt1が生体内で新たなメチル化模様の書き込みに寄与している可能性があると同時に、de novo型のDNAメチルトランスフェラーゼもメチル化模様の維持に貢献していると考えられる。
5.DNA脱メチル化活性
20年も前から多くの研究者により研究されてきた大きな課題に、DNAの脱メチル化反応がある。DNAのメチル化が生理的に重要な意味をもつためには、書き込まれたメチル化模様が消去する過程が存在して、それが積極的に制御されている必要がある。DNAが脱メチル化されるには、複製時にメチル化模様の維持が阻害されるだけでよいことは容易に想像できる。メチル化模様の阻害された状態で2回複製を経ると、半分の細胞では完全に脱メチル化されることになる。胚発生時に起きる脱メチル化はこの機構によるらしい。しかしこれとは別に、複製を介さない、積極的な脱メチル化機構が存在することに疑いの余地はない。このメチル化されたシトシンを積極的に脱メチル化する活性は、ニワトリの性ホルモン感受性ヴィテロジェニン遺伝子のプロモーター、骨格筋型アクチンのプロモーター、受精直後の雄性核、Tリンパ球のインターロイキン2のプロモーターなどで確認されている。
このように、複製を経ない積極的な脱メチル化があることは確かだと考えられ、その機構についてもこれまでいくつかの異なる機構が報告されている。動物では幾つかの機構が報告されたが、何れも他の研究室からの追試報告が無く、確定していない。脱メチル化されるシトシン塩基5位のメチル基は非常に安定な共有結合を形成して、簡単にメチル基だけが取り外される反応は考えにくく、塩基全体を取り除きメチル化されていないシトシンと取り代える機構が考えやすい。最近、植物で特定のゲノム領域のDNA脱メチル化を触媒する酵素として、塩基除去をおこなうグリコシラーゼが2種類同定された38,39)。動物ではアフリカツメガエル卵母細胞にDNA脱メチル化活性が存在することが報告され40)、これに関わる因子の一つが同定された41)。この因子はGadd45aと呼ばれ、塩基除去ではなく、エンドヌクレアーゼによるDNA修復機構に関わる因子であった。直接メチル化されたシトシンを認識して切り出す実体の同定が待たれる。DNA脱メチル化機構のさらなる解明は、DNAメチル化研究にとって残された大きな問題である。
6.おわりに
ゲノムのメチル化模様が新規に形成されるとき、メチル化を受ける標的領域がどのように認識されるのかを明らかにすることが、メチル化形成機構を解明する上で今後の大きな課題である。これについては、上述のように、最近いくつかの報告があり、確実に解明されつつある。また、メチル化維持機構については、Dnmt1がどのようにしてヘミメチル化状態のCpGを認識しているのかを解明することが重要であることは間違いない。しかし、これまでDnmt1が一手に責任を負うとされてきたメチル化模様の維持に、de novo型のDNAメチルトランスフェラーゼの助けが必要であるとの知見が報告され、その機構の詳しい解析が必要となってきており、この点についても進展が待たれる。
文献
1) Bird, A.: Genes Dev., 16, 6 (2002).
2) Bell, A. C., Felsenfeld, G.: Nature, 405, 482 (2000).
3) Hark, A. T., Schoenhere, C. J., Katz, D. J., Ingram, R. S., Levorse, J. M., Tilghman, S. M.: Nature, 405, 486 (2000).
4) Reik, W., Dean, W., Walter, J.: Science, 293, 1089 (2001).
5) Qiu, C., Sawada, K., Zhang, X., Cheng, X.: Nature Struct. Biol., 9, 217 (2002).
6) Okano, M., Bell, D. W., Haber, D. A., Li, E.: Cell, 99, 247 (1999).
7) Watanabe, D., Suetake, I., Tada, T., Tajima, S.: Mech. Dev., 118, 187 (2002).
8) Aoki, A., Suetake, I., Miyagawa, J., Fujio, T., Chijiwa, T., Sasaki, H., Tajima, S.: Nucl. Acids. Res., 29, 3506 (2001).
9) Kaneda, M., Okano, M., Hata, K., Sado, T., Tsujimoto, N., Li, E., Sasaki, H.: Nature, 429, 900 (2004).
10) Sakai, Y., Suetake, I., Shinozaki, F., Yamashina, S., Tajima, S.: Gene Expr. Patterns, 5, 231 (2004).
11) Suetake, I., Morimoto, Y., Fuchikami, T., Abe, K., Tajima, S.: J. Biochem., 140, 553 (2006).
12) Bourc'his, M., Bestor, T. H.: Nature, 431, 96 (2004).
13) Hata, K., Okano, M., Lei, H., Li, E.: Development, 129, 1983 (2002).
14) Suetake, I., Shinozaki, F., Miyagawa, J., Takeshima, H., Tajima, S.: J. Biol. Chem., 279, 27816 (2004).
15) Kareta, M. S., Botello, Z. M., Ennis, J. J., Chou, C., Chedin, F.: J. Biol. Chem., 281, 25893 (2006).
16) Smallwood, A., Esteve, P. O., Pradhan, S., Carey, M.: Genes Dev., 21, 1169 (2007).
17) Suetake, I., Miyazaki, J., Murakami, C., Takeshima, H., Tajima, S.: J. Biochem., 133, 737 (2003).
18) Takeshima, H., Suetake, I., Shimahara, H., Tate, S., Ura, K., Tajima, S.: J. Biochem., 139, 503 (2006).
19) Jeddeloh, J. A., Stokes, T. L., Richards, E. J.: Nature Genet., 22, 94 (1999).
20) Zhu, H., Geiman, T. M., Jiang, O., Schmidtmann, A., Chen, T., Li, E., Muegge, K.: EMBO J., 25, 335 (2006).
21) Gibbons, R. J., McDowell, T. L., Raman, S., O'Rourke, D. M., Garrick, D., Ayyub, H., Higgs, D. R.: Nature Genet., 24, 368 (2000).
22) Fan, Y., Nikitina, T., Zhao, J., Fleury, T. J., Bhattacharyya, R., Bouhassira, E. E., Stein, A., Woodcock, C. L., Skoultchi, A. I.: Cell, 123, 1199 (2005).
23) Tamaru, H., Selker, E. U.: Nature, 414, 277 (2001).
24) Lindroth, A. M., Shultis, D., Jasencakova, Z., Fuchs, J., Johnson, L., Schubert, D., Patnaik, D., Pradhan, S., Goodrich, J., Schubert, I., Jenuwein, T., Khorasanizadeh, S., Jacobsen, S. E.: EMBO J., 23, 4286 (2004).
25) Vire, E., Brenner, C., Deplus, R., Blanchon, L., Fraga, M., Didelot, C., Morey, L., Van Eynde, A., Bernard, D., Vanderwinden, J. M., Bollen, M., Esteller, M., Di Croce, L., de Launoit, Y., Fuks, F.: Nature, 439, 871 (2006).
26) Lehnertz, B., Ueda, Y., Derijck, A. A., Braunschweig, V., Perez-Burgos, L., Kubicek, S., Chen, T., Li, E., Jenuwein, T., Peters, A. H.: Curr. Biol., 13, 1192 (2003).
27) Brenner, C., Deplus, R., Didelot, C., Loriot, A., Vire, E., De Smet, C., Gutierrez, A., Danovi, D., Bernard, D., Boon, T., Pelici, P. G., Amati, B., Kouzarides, T., de Launoit, Y., Di Croce, L., Fuks, F.: EMBO J., 24, 336 (2005).
28) Zhou, Y., Grummt, I.: Curr. Biol., 15, 1434 (2005).
29) Kanno, T., Huettle, B., Mette, M. F., Aufsatz, W., Jaligot, E., Daxinger, L., Kreil, D. P., Matzke, M., Matzke, A. J.: Nature Genet., 37, 761 (2005).
30) Chuang, L. S., Ian, H. I., Koh, T. W., Ng, H. H., Xu, G., Li, B. F.: Science, 277, 1996 (1997).
31) Rountree, M. R., Bachman, K. E., Baylin, S. B.: Nature Genet., 25, 269 (2000).
32) Suetake, I., Hayata, D., Tajima, S.: J. Biochem., 140, 763 (2006).
33) Vilkaitis, G., Suetake, I., Klimasauskas, S., Tajima, S.: J. Biol. Chem., 280, 64 (2005).
34) Ushijima, T., Watanabe, N., Okochi, E., Kaneda, A., Sugimura, T., Miyamoto, K.: Genome Res., 13, 868 (2003).
35) Laird, C. D., Pleasant, N. D., Clark, A. D., Sneeden, J. L., Hassan, K. M., Manley, N. C., Vary, J. C. Jr., Morgan, T., Hansen, R. S., Stoger, R.: Proc. Natl. Acad. Sci. USA, 101, 204 (2004).
36) Dodge, J. E., Okano, M., Dick, F., Tsujimoto, N., Chen, T., Wang, S., Ueda, Y., Dyson, N., Li, E.: J. Biol. Chem., 280, 17986 (2005).
37) Kim, G. D., Ni, J., Kelesoglu, N., Roberts, R. J., Pradhan, S.: EMBO J., 21, 4183 (2002).
38) Morales-Ruiz, T., Ortega-Galisteo, A. P., Ponferrada-Marin, M. I., Martinez-Macias, M. I., Ariza, R. R., Roldan-Arjona, T.: Proc. Natl. Acad. Sci. USA, 103, 6853 (2006).
39) Agius, F., Kapoor, A., Zhu, J. K.: Proc. Natl. Acad. Sci. USA, 103, 11796 (2006).
40) Simonsson, S., Gurdon, J.: Nature Cell Biol., 6, 984 (2004).
41) Barreto, G., Schafer, A., Marhold, J., Stach, D., Swaminathan, S. K., Handa, V., Doderlein, G., Maltry, N., Wu, W., Lyko, F., Niehrs, C.: Nature, 445, 671 (2007).
![]()