【トピックス】
クロロアルコール脱ハロゲン化酵素と光学分割への応用
中川 篤、鈴木利雄
ダイソー・研究所
1.はじめに
光学活性化合物の創製において、光学的に高純度かつ化学的に純粋な合成ブロックを出発原料とすることは普遍的な方法であり、効果的な方法である。自然界にある糖質やアミノ酸からの合成方法は良く知られているが、一方の鏡像体しかない場合や炭素数に制限があるなどいくつかの問題が知られていた1)。我々はこれらのニーズに応えるべく、既にクロロプロパノールやクロロプロパンジオールの微生物資化分割法により、C3キラル化合物として代表される光学活性エピクロロヒドリン、グリシドール、そしてグリシジル誘導体などの汎用性の高い光学活性C3合成ユニットの生産について研究を進めてきた2)。これらの光学活性C3合成ユニットはもっとも簡単なキラル化合物であり、単純な糖質でもある炭素数3のグリセロール骨格を有し、反応性の高いエポキシ基や塩素原子あるいは利用し易い脱離基やその他の官能基を有しているため広範囲な応用が可能である。
さらに我々は光学活性C4合成ユニットとして有用な4-クロロ-3-ヒドロキシブチロニトリル (BN)、4-クロロ-3-ヒドロキシブタン酸エステル (CHB) や3-ヒドロキシ-γ-ブチロラクトン (HL) の微生物休止菌体法による光学分割法の開発を進めてきた2)。これらの光学活性C4合成ユニットはβ-ヒドロキシ酸類やL-カルニチンの合成の出発化合物として重要である。特に(S)-BNや(S)-CHBは、最近では抗高脂血症剤として効果のあるHMG-CoAリダクターゼ阻害剤であるスタチン系の薬剤の中間体合成に利用されている。
そこで、これらのC3ならびにC4ハロアルコールの光学分割に関与する酵素のうち、3-クロロ-1,2-プロパンジオールとCHBの立体選択的脱ハロゲン化酵素に関する検討を行った。本総説では、我々が研究を進めてきたクロロプロパンジオールならびにクロロヒドロキシブタン酸エステルの立体選択的な脱ハロゲン化酵素に関して、酵素化学的な諸性質を明らかにするとともに、遺伝子レベルでの解析について紹介する。さらにそれらを用いた光学活性1,2-ジオールおよびC4合成ユニット開発についても述べる。
2.クロロプロパンジオール脱ハロゲン化酵素について
2-1 3-クロロ-1,2-プロパンジオール (CPD) の立体選択的光学分割について
我々は、効果的な光学活性合成ユニットの開発を目指して研究に着手した結果、汎用性の高い光学活性C3合成ユニットの1つである光学活性CPDの微生物培養法による光学分割法を開発した。土壌より単離された微生物はPseudomonas属とAlcaligenes 属に属する細菌で、本微生物によるCPDの光学分割の原理は、ラセミ体CPDのうち一方の鏡像体のみを立体選択的に分解資化することに基づいている (立体選択的資化分割法)3,4) (図1)。立体選択的資化法とは、ラセミ体からなる化合物を炭素源とし、必要の無い鏡像体は微生物により炭素源として利用・分解され、微生物の生育を伴うことを特徴としている。その結果、培養終了後の培養液中には必要な鏡像体と生育・増殖した微生物菌体しか残存しないため、回収工程は容易に行うことができるという利点がある。我々は1994年に松山工場において本技術に基づく光学活性CPDの工業的生産を開始した。
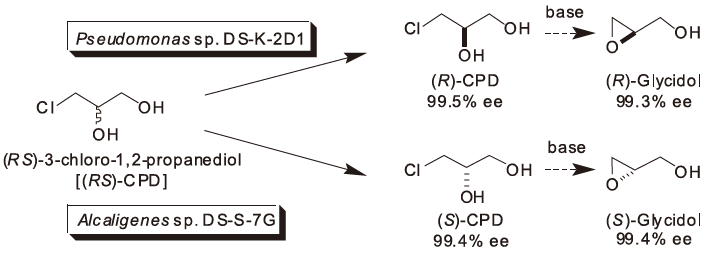
図1 微生物を用いた立体選択的資化法による光学活性CPDの生産
ハロゲン化低級脂肪族炭化水素の微生物分解は古くから知られている5)。一方、クロロプロパノールやクロロプロパンジオールに代表されるハロアルコールの脱ハロゲン化酵素については、現在数多くの研究が報告されているが6)、古くはCastroら7,8)やJanssenら9,10)による報告が知られていた。そのほとんどはHalohydrin halide hydrogen lyaseに属し、ハロアルコールを脱クロル化しながらエポキシドへと変換する酵素 (エポキシダーゼ) がほとんどであった11)。
我々は、本資化分割に用いた細菌の立体選択的ハロアルコール脱ハロゲン化酵素について、その基質特異性や酵素化学的な性質の検討を行った結果、(R)-CPD資化細菌であるAlcaligenes sp. DS-S-7G株由来の酵素は、アルコールデヒドロゲナーゼ様の活性を伴いながら脱クロル化反応を触媒するという興味深い活性を示した。本項では、(R)-CPD資化細菌であるAlcaligenes sp. DS-S-7G株の脱ハロゲン化酵素の反応メカニズムについて述べ、さらに本ハロアルコール脱ハロゲン化酵素による光学活性1,2-ジオール合成ユニット生産への応用について紹介する。
2-2 新規な(R)-CPD脱ハロゲン化酵素の分離、精製とその性質
ラセミ体CPDを単一炭素源とする合成培地で、(R)-CPD資化性細菌Alcaligenes sp. DS-S-7G株を培養し、菌体を超音波破砕後、種々のクロマトグラフィーにより(R)-CPD脱ハロゲン化酵素の分離、精製を行い、そのメカニズムを検討した12)。その結果、(R)-CPD脱ハロゲン化反応に関与する酵素は2種類のタンパクが関与していることが明らかとなり、それぞれEnzyme 1 (分子量70 kDa, ヘテロ二量体16 kDa and 58 kDa、1分子のFAD を非共有的に結合)、Enzyme 2 (分子量86 kDa, 33 kDa and 53 kDa、ヘテロ二量体) と命名した。本脱ハロゲン化酵素による反応メカニズムはNAD+を補酵素とする酸化的脱ハロゲン化反応であることが判明した。また好気的条件下においてEnzyme 1単独でも脱ハロゲン化活性を触媒し、ハイドロキシアセトンを生成した。Enzyme 2との共役作用によりその活性は4~5倍に上昇するという興味ある知見を得た。一方、Enzyme 2のみではCPD脱ハロゲン化活性を示さなかった (図2)。これよりEnzyme 2の脱ハロゲン化反応における役割はEnzyme 1中のFADからNAD+への電子伝達あるいはその調節に関与していると考察した。また、Enzyme 1に対しDCIP (2,6-dichlorophenolindophenol) とPMS (phanazine methosulfate) は、NAD+にかわる最適な電子受容体になることが判明し、その活性は1,000倍に上昇した。このような立体選択的かつ酸化的な脱ハロゲン化酵素の報告は知られておらず、そこでこの活性を有するEnzyme 1をハロアルコール酸化的脱ハロゲン化酵素 (halohydrin dehydro-dehalogenase; HDDase) と命名した。
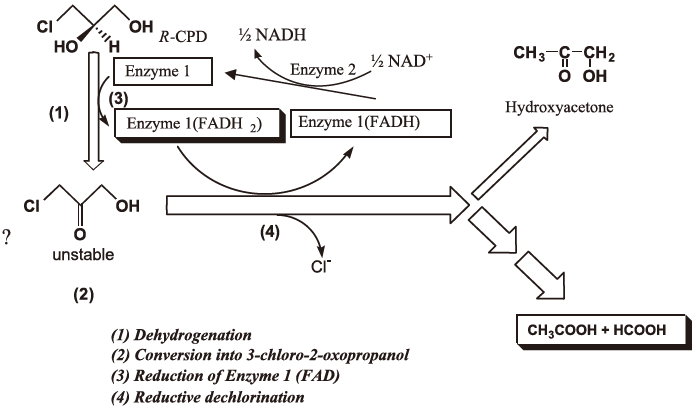
図2 予測される(R)-CPD脱ハロゲン化メカニズム
2-3 (R)-CPD脱ハロゲン化酵素遺伝子のクローニング
本菌株からHDDase (Enzyme 1) およびEnzyme 2をコードする遺伝子 (HDD、E2) を上述した精製酵素の部分アミノ酸配列情報をもとに、コロニーハイブリダイゼーション等の常法を用いてクローニングした13,14)。得られたDNA断片の塩基配列を決定した結果、図3に示すようにHDD、E2遺伝子のORFは並んでおり、順にEnzyme 2の大サブユニット、HDDaseの大サブユニット、同小サブユニットをコードする1437、1629、471bpの各ORFが確認でき、各々E2L、HDD-L、HDD-S遺伝子と命名した。また、Enzyme 2の小サブユニットをコードするORFは確認できなかった。精製したAlcaligenes sp. DS-S-7G株のEnzyme 2の大サブユニットと小サブユニットのN末端アミノ酸配列が同一であったことから、Enzyme 2の小サブユニットはE2L遺伝子発現後のプロセッシングにより形成されることが考えられる。また、HDD-LとHDD-S遺伝子はオペロンを形成していることが判明した。塩基配列から推定されるアミノ酸配列より、HDDaseは種々微生物のグルコン酸脱水素酵素、ソルビトール脱水素酵素等と相同性を示し、既に報告されているハロアルコールの脱クロル化酵素とは異なることが示唆された。また、大サブユニットのN末端側には、FAD結合部位の存在が確認され、酸化的な触媒作用を有することも証明された。一方、Enzyme 2は微生物各種のアルデヒド脱水素酵素と高い相同性を示した。このように、推定アミノ酸配列からも本酵素がCPD資化の一連の酸化反応を触媒することが示唆された。現在、発現タンパクを用いてHDDaseとEnzyme 2の共役作用の詳細解析を行っている。
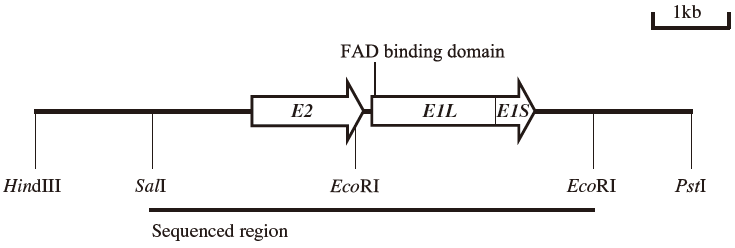
図3 (R)-CPD脱ハロゲン化酵素遺伝子群を含むクローニング断片
2-4 HDDase酵素遺伝子 (HDD) の発現
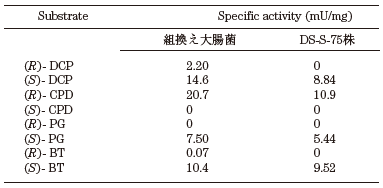
HDDaseが立体選択的脱ハロゲン化活性を触媒する主酵素であることから、HDD遺伝子をプラスミドベクターpUC18のLacプロモーター支配下に挿入し、大腸菌JM109株に導入した。得られた組換え大腸菌をLB培地で、16時間、30℃で好気的に培養し、対数増殖期にIPTGによる誘導を行うことにより、HDD遺伝子を高発現させた。この細胞抽出液についてNative-PAGE 後に活性染色14)を行った結果、ゲル上の同じ位置にAlcaligenes sp. DS-S-7G株由来のHDDase細胞抽出液の活性染色バンドが検出されたことから、HDD遺伝子を導入した組換え大腸菌はAlcaligenes sp. DS-S-7G株と同様にHDDase活性を保持していることが確認された。さらに、光学活性DCP、CPD、1,2,4-ブタントリオール (BT) や1,2-プロパンジオール (PG) などの1,2-ジオールの各基質に本組換え大腸菌の細胞抽出液を反応させた結果、後述のように各基質に対してHDDaseと同一の立体選択性を有した (表1)。現在、残るE2遺伝子の発現を検討しており、HDD遺伝子との共発現、さらに、NAD+や電子受容体等の補酵素再生系を発現タンパク質レベルで行うことで、より高い生産性を得ることを視野に入れて開発を行っている。
表1 HDD遺伝子を導入した組換え大腸菌由来酵素の活性
3.光学活性1,2-ジオール合成ユニットの生産
3-1 光学活性1,2-ジオール合成ユニットについて
光学活性1,2-ジオール合成ユニットは、医薬・農薬や新素材の分野における光学活性ユニットとしての利用が期待される有用な化合物である。中でも光学活性1,2 -プロパンジオール (PG) は、光学活性C3合成ユニットの一つとして重要な化合物で、(R)-PGは抗エイズ薬 (Viread) や抗菌剤 (Levofloxacine) の合成に用いられている15,16)。光学活性PGの実用的な生産法としては,野依らが開発したBINAP触媒を用いるヒドロキシアセトンの不斉還元法17)が高砂香料において、またJacobsen らが開発した光学活性salen-Co (III) 錯体を用いたプロピレンオキシドの速度論的光学分割法18)がRhodia において工業化された。そこで我々は,より経済的かつ技術的に競争力のある製法を求めてその生産法の開発に注力した。
3-2 HDDaseを利用した光学活性1,2-ジオール合成ユニットの調製
我々の方法の特徴は、(R)-CPD資化細菌であるAlcaligenes sp. DS-S-7G株の立体選択的脱ハロゲン化メカニズムを解明中に発見単離したHDDaseを用いている点である。本酵素は、(R)-CPDや(S)-2,3-ジクロロ-1-プロパノール (DCP) などのハロアルコールに高い脱クロル化活性を示すだけでなく、PG を始めC3~C6の種々の1,2-ジオール化合物についても高い立体選択性を示し、酸化的に相当するアルデヒドを経由してギ酸にまで分解する。その結果、ハロゲン化あるいは非ハロゲン化光学活性1,2-ジオールを高光学純度、高収率で簡便に調製することができた19,20) (図4)。すなわち我々のHDDaseを用いる方法は新規な光学活性1,2-ジオール合成ユニットの調製法とも考えられ、今後、さらに補酵素であるNAD+や電子受容体の効果的なリサイクルが可能になれば、工業的な方法の1つであると考えられる。既述の遺伝子組換え法を用いた技術により、本酵素の大量生産が可能になれば安価で工業的な光学活性1,2-ジオール合成ユニットの生産法になると考えられる。
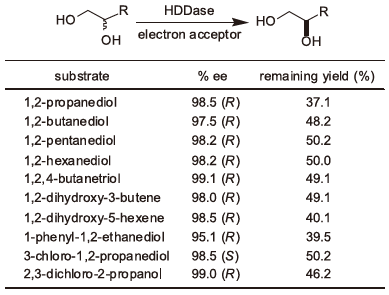
図4 HDDaseによる光学活性1,2-ジオール合成ユニットの創製
4.光学活性C4合成ユニットの開発
4-1 光学活性C4合成ユニットの有用性
光学活性C4合成ユニットの開発について紹介する。光学活性C4合成ユニットは光学活性C3合成ユニットと同様に医薬・農薬・天然化合物や新素材へと展開できる点で重要な化合物である。β-ヒドロキシブタン酸や1,3-ブタンジオールなどの光学活性C4化合物は、カルバペネム系抗生物質などに代表される抗菌剤合成に利用されている21)。図5に示すように4-クロロ-3-ヒドロキシ-ブタン酸エステル (CHB) と4-クロロ-3-ヒドロキシブチロニトリル (BN) はL-カルニチン、L-GABOB、β-ヒドロキシ酸、3-ヒドロキシ-γ-ブチロラクトン (HL)、4-ヒドロキシ-2-ピロリドンなどの有用な化合物への展開が可能な光学活性C4合成ユニットである。最近では抗高脂血症薬として有用なHMG-CoAリダクターゼ阻害剤などの中間体22)としても重要な化合物でもある。
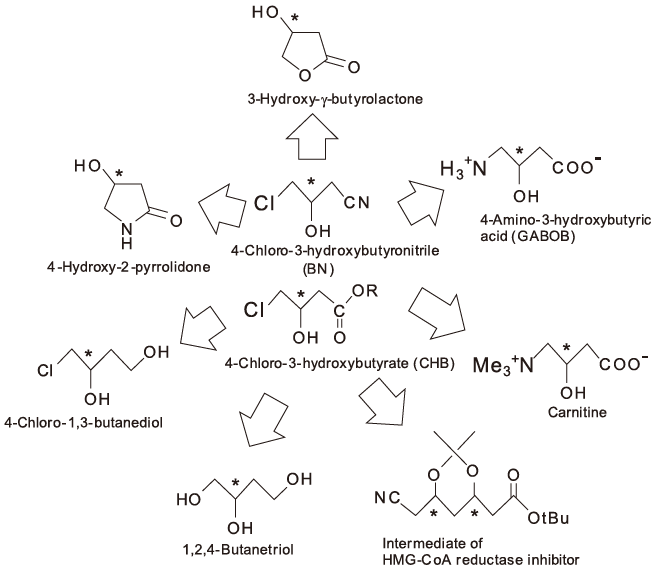
図5 光学活性C4合成ユニットの応用
これらの光学活性BNやCHBはそれらの構造上、両化合物とも有機合成化学的には光学活性EPよりシアン化カリウムを付加させてアルコリシスなどにより誘導することは可能であるが、コスト面からさらに川下で分割する方法を検討した。すでにHalohydrin halide-hydrogen lyase を利用する(R)-BN の生産法23)やカルボニルレダクターゼ21,24)やBINAP触媒17)による不斉還元法を用いた光学活性CHBの生産が報告されている。しかしながら、一方の光学活性体しか得られなかったり、光学純度が低かったり (BINAP触媒を用いた場合94%ee)、高価な触媒や補酵素 (NADPHやNADH) を必要とするなど、工業的生産の観点から種々の課題が残されていた。
4-2 光学活性C4合成ユニットの微生物光学分割
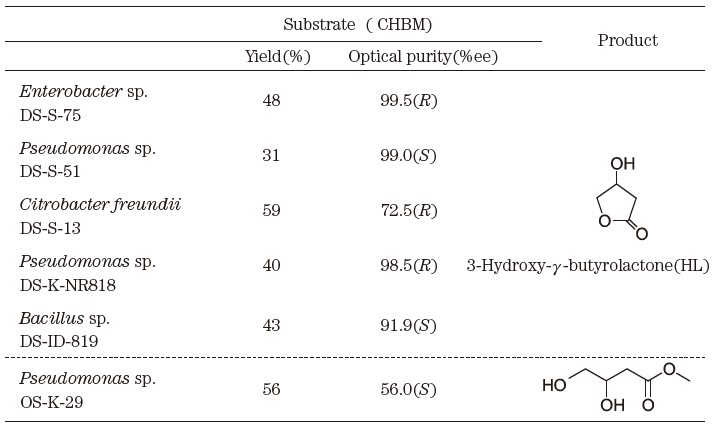
我々は、土壌より単離した微生物を用いて種々のハロアルコール化合物より光学活性C4合成ユニットの創製について検討した25)。ハロアルコール化合物を立体選択的脱クロル化反応によりジオールへと加水分解する反応を触媒するPseudomonas属細菌の脱クロル化酵素をラセミ体BNに作用させたところ、(R)-BN を脱クロル化しながら相当する(R)-ジオールに変換し、反応液中に(S)-BNを94.5%ee、収率35-40%で得ることができた。また、(S)-BN 生産と同様に(S)-CHB生産にも応用できることが判明した。さらに光学活性C4合成ユニット分割生産微生物のスクリーニングを実施した結果、新たにPseudomonas属、Enterobacter 属、Citrobacter属、Bacillus属細菌を単離した。これらの微生物は、CHBを立体選択的脱ハロゲン化活性により、一方の光学異性体CHBを相当するHLに変換した (表2)。
表2 光学活性C4合成ユニット生産微生物のスクリーニング
次いで、立体選択性の高かったEnterobacter sp. DS-S-75株の休止菌体を用いたラセミ体CHBの光学分割反応についてさらに検討を進めた結果、本休止菌体はCHBに対して図6に示すような反応を触媒することを明らかにした26)。すなわち本反応では非常に高い立体選択的脱クロル化反応が進み(S)-CHBは(S)-HLへと変換された。最終的には反応液中に(R)-CHBと(S)-HLが残存し、それぞれ収率48%で>99%ee の(R)-CHBと90%eeの(S)-HLをone potで生産することができた。DS-S-75株の休止菌体は全く(R)-CHBに対して脱クロル化活性を示さず、本光学分割反応は完全な立体選択性の下で反応が進んでいることが示唆された。次いでその立体選択性をCHBのメチル、エチル、プロピル、イソプロピル、ブチル各エステルに対して比べたところ、そのメチルCHB (CHBM) に対して一番高い活性を示した。
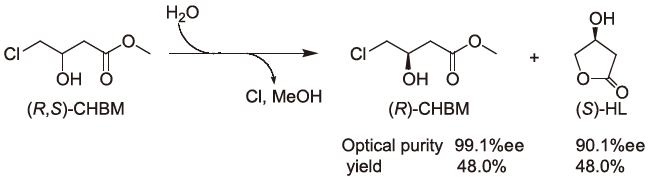
図6 Enterobacter sp. DS-S-75株によるラセミ体CHBの光学分割
(S)-HLはL-リンゴ酸やD-あるいはL-ヘキソースからの合成法27,28)が報告されていたが、我々は新規な立体選択的脱ハロゲン化活性を有するEnterobacter属細菌を用いることにより(S)-HLを生産できるようになった。しかしながら、いずれのCHBの場合も得られる(S)-HL の光学純度が低いなど、さらなる光学分割法の改良が求められた。
4-3 光学活性C4合成ユニットの生産とその脱ハロゲン化酵素について
先に述べたように、Enterobacter sp. DS-S-75株を用いる休止菌体反応によりラセミ体CHBMの光学分割反応法を確立し、同時にその生成物である(S)-HLの生産法も可能にした。しかし工業的生産を行なうために更なる活性改良や立体選択性および基質特異性の改良が必要となった。そこで、我々は本分割反応に関与する脱ハロゲン化酵素の単離精製とその諸性質を明らかにした29)。
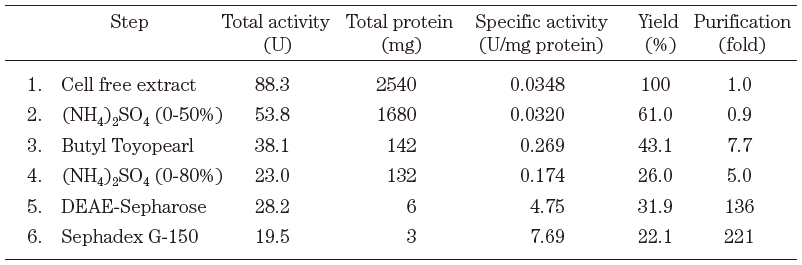
DS-S-75株の菌体破砕液を脱クロル活性を指標30)に各種カラムクロマトグラフィーに供した結果、SDS-PAGE31)で単一バンドになり、221倍まで精製した (表3)。ゲルろ過カラムによる分子量は75 kDaであり、SDS-PAGEの結果も合わせると、37.5 kDaのサブユニットからなるホモ二量体であることがわかった。本酵素の諸性質を解析した結果、(S)-CHBMに対するKm値は8.04 mM、Vmaxは19.2 U/mg、pH5.0-8.5で安定であり、最適pHは6.6-6.8、金属の要求性はなかった。精製酵素の基質特異性を調べた結果、CHB等のβ-ヒドロキシ酸、およびα-ヒドロキシ酸などの各種カルボン酸エステルに加水分解活性を示した。しかしながら、BNやCPD等のハロアルコールやジエステルに対しては分解活性を示さなかった。これらの基質特異性から、本酵素はカルボン酸エステル水解活性を有していることが示唆された。
表3 DS-S-75株由来CHB脱ハロゲン化酵素の精製
4-4 CHB脱ハロゲン化酵素遺伝子の単離と高発現
そこで(R)-CHBの生産性をさらに向上させるべく、組換え体を用いた光学分割反応を行うことを目的として、精製酵素から決定したアミノ酸配列情報から、先述の(R)-CPD脱ハロゲン化酵素遺伝子のクローニングと同様にコロニーハイブリダイゼーション法等の常法により、DS-S-75株から本酵素遺伝子を含むDNA断片を単離した。この塩基配列を決定したところ、1101bp、367アミノ酸をコードするORFが存在し、想定されるアミノ酸配列中にDS-S-75株から精製した酵素と同一のアミノ酸配列が存在した。精製酵素のN末端アミノ酸配列はバリンであり、開始メチオニンから二十数残基内部にあった (図7)。配列中にリパーゼ、エステラーゼなどの加水分解酵素に保存されているGXSXGからなるアミノ酸配列が存在し、微生物各種のカルボキシエステラーゼ、リパーゼと相同性が見られたが、最大で45%と低かった。これらの結果からも(S)-CHB脱ハロゲン化酵素は、カルボン酸エステル分解酵素であることが明らかとなった。
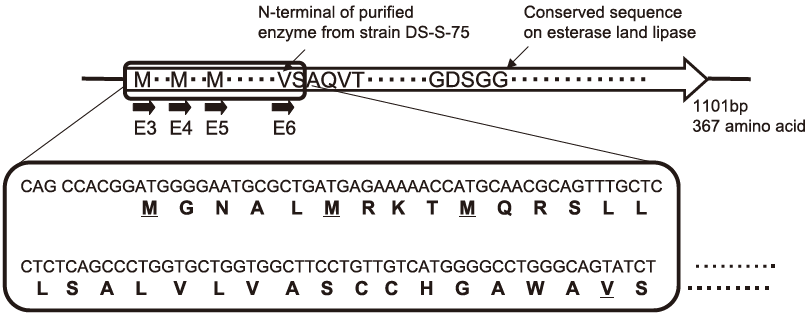
図7 CHB脱ハロゲン化酵素遺伝子と推定アミノ酸配列
配列中に翻訳開始のメチオニンをコードするコドンと考えられるATG配列が3箇所存在した。各配列から翻訳を開始させるよう、EcoRI 部位を付加したPCRプライマーを設計し、PCR産物をpKK223-3 のEcoRI-PstI 部位に導入した。また、精製酵素のN末端のバリンすなわち成熟酵素のN末であるバリンもメチオニンに変換して同様に導入した。構築したプラスミドを大腸菌JM109およびDH5α株に導入し、IPTGによる誘導なしにLB培地にて16時間振とう培養した培養液を用いて、p-ニトロフェニルブタン酸エステルを基質として、形質転換体の加水分解活性を測定した。表4は菌株、培養液のOD、培養液あたりの活性、培養液あたりの比活性を示す。1分間に1µ mol分解させる酵素量を1Uと定義した。JM109 (pKK-E4) が活性、比活性とも高く、また、すぐ上流にSD配列と想定されるGAに富んだ配列が存在することから、本来の開始メチオニンであると示唆される。一方、E5、E6の活性は低く、N末端の25残基からなるアミノ酸配列は酵素の安定化に関与していることが示唆された。また、SDS-PAGEではDS-S-75株由来の精製酵素と同じ37.5 kDa の位置にバンドが確認され (図8)、組換え酵素のN末端アミノ酸配列を10残基解析したところ、DS-S-75株由来の精製酵素のN末端配列と一致した。また、ゲルろ過カラムクロマトグラフィーによる分子量も75 kDa と一致した。これらの結果より、組換え酵素も大腸菌内でN末端配列がプロセッシングされ、二量体を形成していることがわかった。
表4 DS-S-75株および組換え大腸菌培養液の活性
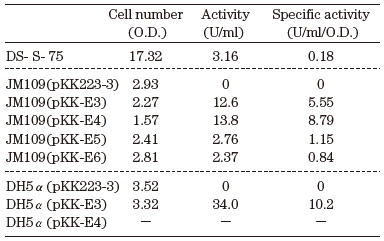

図8 精製酵素および組換え大腸菌抽出液のSDS-PAGE
JM109 (pKK-E4) は菌体量が他よりも少なく、高発現による生育阻害が考えられ、IPTGによる誘導を行なうと、全く生育しなかった。また、構成的に発現させるためにDH5α32)を宿主に用いた場合は形質転換体を得ることができなかった。JM109 (pKK-E3) は生育阻害が少なく、DH5αを宿主に用いた場合、pKK-E3は構成的に発現し、培養液あたりの活性は、親株DS-S-75 株の約10 倍になった。形質転換体の工業的利用を考慮した場合、誘導剤なしに構成的にかつ安定に高発現することは好都合であり、以後このDH5α (pKK-E3) について検討を行った。
4-5 遺伝子組換え大腸菌を利用したCHBMの光学分割
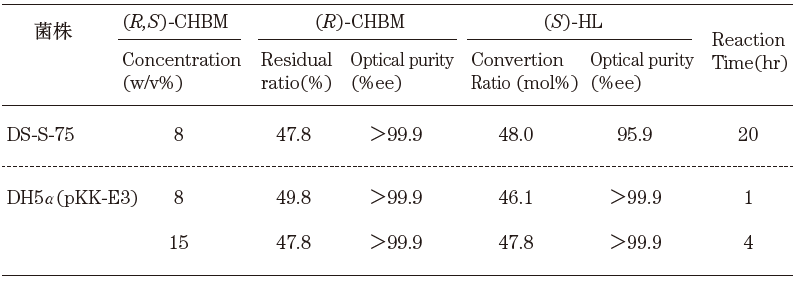
作製したDH5α (pKK-E3) を用いてCHBMの光学分割反応を行なった。2%ペプトン、1%酵母エキス、0.5% グリセリン、0.01% アンピシリンからなる栄養培地にて30℃で培養後、培養液にラセミ体CHBMを添加し、30℃で中和剤にアンモニア水を用いて反応させた。その結果、8%濃度のCHBMを完全に分割するまでDS-S-75株は20時間を要していたが、形質転換体では、高活性であるため1時間で分割することができた。さらに15% 濃度のCHBM も分割することができ、反応1バッチあたりの生産量が増加した。また、生成するS体HLの光学純度も99%ee以上であった (表5)。形質転換体を用いることで目的酵素のみを大量発現することができたため、立体選択性が向上したと考えられる。光学活性HLもまた医薬中間体として有用な化合物であり、これまでに多くの製法が開発されている。本光学分割反応は、培養液をそのまま生体触媒として利用可能で、高価な補酵素を必要とせず、非常に立体選択性の高いカルボン酸加水分解酵素1種により温和な条件下で触媒されるため、工業的利用に非常に有効である。
表5 CHBMの光学分割
さらに本酵素の立体選択的な基質特異性から、α-およびβ-ヒドロキシ酸やテトラヒドロフラン-2-カルボン酸の各エステル等の光学分割が可能になった2)。現在、詳細な研究とその遺伝子に関する研究が進められており、その遺伝子を用いた工業的な生産法が検討されている。
5.おわりに
我々は立体選択的にラセミ体C3ハロアルコールを資化分割し、さらにはラセミ体C4ハロアルコールを立体選択的な生体触媒により変換することにより有用な光学活性C3、C4合成ユニットであるクロロプロパンジオールや1,2-ジオール類、さらにはCHBおよびHLの生産に成功した。そして現在では70種類以上ものそれらの誘導体を供給することができる。我々の方法はラセミ体のうち一方の鏡像体しか与えることはできないが、工業的な生産においては分割されるいずれのラセミ体も石油化学工業において大量安価に供給されているため、経済的に有利な方法の一つであると考えられる。そして、我々が開発した光学活性C3,C4合成ユニットは医薬などのファインケミカル分野を中心に利用されており、そのニーズはさらに大きくなるものと考えている。将来に向けて、微生物、酵素を利用した生化学的な技術と有機化学的技術が融合し補いながらこのようなキラルプロセス化学を発展させていくことが重要であろう。
謝辞
本研究を遂行するにあたり貴重なご助言とご提案を頂きまし多くの研究者の皆様に感謝致します。またこれらC3、C4キラル合成ユニットの生産研究の発展にご支援いただきましたダイソー株式会社の皆様に感謝致します。
文献
1) Crosby, J., Collins, G. N., Sheldrake, G. N.: Chirality in Industry (Crosby, J. ed.) pp.5-20, John Wiley & Sons (1992).
2) Kasai, N., Suzuki, T.: Adv. Synth. Catal., 345, 437 (2003).
3) Suzuki, T., Kasai, N., Yamamoto, R., Minamiura, N.: J. Ferment. Bioeng., 73, 443 (1992).
4) Suzuki, T., Kasai, N., Minamiura, N.: Appl. Microbiol. Biotechnol., 40, 273 (1993).
5) Slater, H., Bull, A. T., Hardman, D. J.: iodegradation, 6, 181 (1995).
6) Kasai, N., Suzuki, T., Furukawa, Y.: J. Mol. Cat. B: Enzymatic, 4, 237 (1998).
7) Castro, C. E., Bartnicki, E. W.: Biochemistry, 7, 3213 (1968).
8) Bartnicki, E. W., Castro, C. E.: Biochemistry, 8, 4677 (1969).
9) Wijngaard van den, A. J., Janssen, D. B., Witholt, B.: J. Gen. Microbiol., 135, 2199 (1989).
10) Wijngaard van den, A. J., Reuvekamp, P. T. W., Janssen, D. B.: J. Bacteriol., 173, 124 (1991).
11) Nakamura, S., Nagasawa, T., Yu, F., Watanabe, I., Yamada, H.: J. Bacteriol., 174, 7613 (1992).
12) Suzuki, T., Kasai, N., Yamamoto, R., Minamiura, N.: Appl. Microbiol. Biotechnol., 42, 270 (1994).
13) 中川 篤、鈴木利雄、加藤 晃、新名惇彦、WO2003/018796.
14) 鈴木利雄、中川 篤、西川孝治:ファインケミカル, 36, 52 (2007).
15) Schultze, L. M., Chapman, H. H., Dubree, N. J. P., Jones, R. J., Kent, K. M., Lee, T. T., Louie, M. S., Postich, M. J., Prisbe, E. J., Rohloff, J. C., Yu, R. H.: Tetrahedron Lett., 39, 1853 (1998).
16) Ebata, T.: Process Technology for Intermediate of Chiral Pharmaceuticals (Shinkai, I. ed.), pp.150-155, Gijutsujohokyokai, Tokyo.
17) Kitamura, M., Tokunaga, M., Ohkuma, T., Noyori, R.: Tetrahedron Lett., 32, 4163 (1991).
18) Tokunaga, M., Larrow, J. F., Kakiuchi, F., Jacobsen, E. N.: Science, 277, 936 (1998).
19) Suzuki, T., Kasai, N., Minamiura, N.: Tetrahedron: Asymmetry, 5, 239 (1994).
20) Suzuki, T., Kasai, N., Minamiura, N.: J. Ferment. Bioeng., 78, 194 (1994).
21) Matsuyama, A., Yamamoto, H., Kobayashi, Y.: Org. Proc. Res. & Develop., 6, 558 (2002).
22) Wolberg, M., Hummel. W., Wandrey, C., Muller, M.: Angew. Chem. Int. Ed., 39, 4306 (2000).
23) Nakamura, T., Nagasawa, T., Yu, F., Watanabe, I., Yamada, H.: Biochem. Biophysic. Res. Commun., 180, 124 (1991).
24) Zhou, B., Gopalan, A. S., VanMiddlesworth, F., Shieh, W.-R., Sih, C. J.: J. Am. Chem. Soc., 105, 5925 (1983).
25) Suzuki, T., Kasai, N.: Trends Glycosci. Glycotechnol., 15, 329 (2003).
26) Suzuki, T., Idogaki, H., Kasai., N.: Enzyme. Microb. Technol., 24, 13 (1999).
27) Inoue, K.,: J. Syn. Org. Chem. Jpn., 59, 430 (2001).
28) Hollingsworth, R. I.: J. Org. Chem., 64, 7633 (1999).
29) Nakagawa, A., Idogaki, H., Kato, K., Shinmyo, A., Suzuki, T.: J. Biosci. Bioeng., 101, 97(2006).
30) Iwasaki, I., Utsumi, S., Ozawa, T.: Bull. Chem. Soc. Jpn., 25, 226 (1952).
31) Leammli, U. K.: Nature, 227, 680 (1970).
32) Grant, S. G. N., Jessee, J., Bloom, F. R., Hanahan, D.: Proc. Natl. Acad. Sci. USA, 87,4645 (1990).
![]()