【トピックス】
ヘム酵素の創成
渡辺芳人、荘司長三
名大院・理
1.はじめに
分子生物学が強烈な勢いで発展し、クローン、ES細胞や幹細胞などの言葉が一般のニュースでも日常的に聞かれるようになって久しい。我々化学者も、遺伝子操作によってタンパク質の一部のアミノ酸を置き換えたミュータントタンパク質は、もはや特別な世界の存在ではなくなっている。しかしながら、「有機化学や無機化学の研究室で自前のミュータントを作成し、研究材料に使う」となると、ちょっと変わったことをしている研究室ということになるのが普通であろう。不斉触媒の開発は、有機合成分野では日常的に行われている研究である。一方で、酵素は、我々の体の中で様々な不斉合成を触媒していることはみんなが知っている。さらに、酵素の触媒活性中心構造を合成化学的に作りだし、触媒研究に役立てるbiomimetic化学は、30年近い歴史を持っている。また、現実に存在する酵素を合成反応に利用する研究は多数あり、不斉加水分解反応を触媒するリパーゼなどはその代表格と言える1)。この反応は可逆反応なので、アルコールとカルボン酸が存在する条件では、エステル合成も可能となる。しかしながら、非天然基質に対してリパーゼがどのような特異性・選択性で加水分解反応を触媒するかは、スクリーニング的手法で網羅的に調べる必要があり、カスタムメイドの触媒というわけにはいかない。そこで、「我々が希望する反応を触媒する酵素」を作り上げる方法論や技術が開発されれば、一般の化学者も酵素化学の分野にこぞって参入すると期待される。
さて、私の研究室では、長年にわたって酸化反応を触媒するヘム酵素の研究を行っている。代表的なヘム酵素として、シトクロムP450、西洋わさびペルオキシダーゼ (HRP) やシトクロムcペルオキシダーゼ (CcP)、カタラーゼがよく知られている (表1)2)。ヘム酵素は、活性中心 (触媒中心) がヘムと呼ばれる鉄ポルフィリン錯体によって構成されている酵素の総称であり、タンパク質を使った研究以外に、合成金属ポルフィリン錯体を酵素のモデル錯体として利用した膨大な研究が行われている3)。著者らは、タンパク質を使った酸化反応から研究に入り (1970年代)4)、活性種のキャラクタリゼーションとその生成機構を研究するために、モデル錯体を使った研究へと順番にシフトしていった (1980年代)5)。モデル系による活性種生成機構の詳細が分かった段階で、「タンパク質の中でも同じ反応が進行しているのだろうか?」という興味が湧き始め、再び酵素自体を対象にする研究へと戻ることにした (1990年代)。
表1 ヘム蛋白質・酵素の代表的な機能

90年代の酵素研究は、分子生物学の興隆によって70年代の研究とは様変わりである。70年代は、ウサギの肝臓から単離した酵素を使っていたが、90年代には、遺伝子工学的に大腸菌で作らせた酵素を使い、必要に応じて特定の部位にあるアミノ酸を置き換えたミュータントも自在に作成できる段階に入っていた。
2. 酵素設計の第一歩:ミオグロビンミュータント
ヘム酵素は、表1に示すように多くの酸化反応に関与している。その中でも、ペルオキシダーゼは魅力的な酵素といえる。その理由は、ペルオキシダーゼが過酸化水素を酸化剤として用いているところにある。大規模な物質生産を考えるとき、酸化剤が安価であり、反応後の副生成物がH2Oのみであることは、非常に重要なポイントとなる。
さて、酸素分子の運搬や貯蔵を受け持つヘムタンパク質であるヘモグロビン (Hb) やミオグロビン (Mb) も、活性中心は鉄ポルフィリンが構成している。HbやMbの触媒中心の近傍構造は、CcPのようなペルオキシダーゼに似ているにもかかわらず (図1)、酸化反応を触媒することは出来ない。構造的に類似しているにもかかわらずその化学的な機能 (反応性) が異なるというからには、そこには反応論的な理由があるはずである。そういう目で図1の構造を比較すると、鉄イオンとヒスチジン (His) との距離がMbとCcPでは、1Åほど異なる事に気がつく。
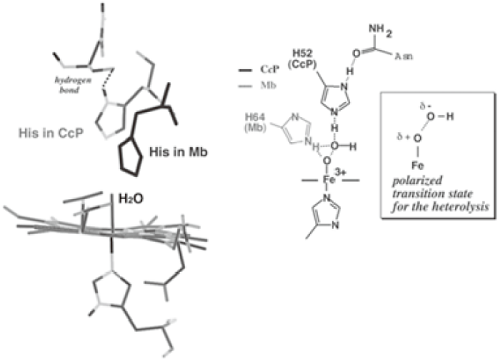
図1 MbとCcPのヘム近傍構造の比較
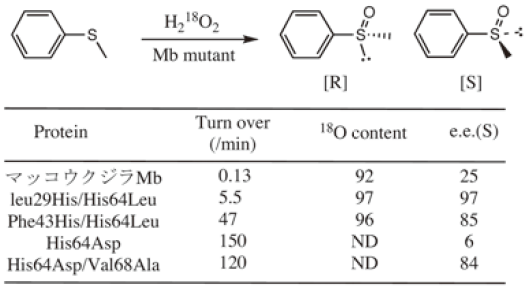
ヘム酵素による酸化反応では、高原子価状態の酸化活性種としてcompound Iと呼ばれるO=Fe(IV) Porphyrin (Por)+•の生成が重要である。過酸化水素とペルオキシダーゼの反応を例に取ると、図1(右) に示すように、鉄に結合した過酸化水素はFe--O(δ+)--O(δ–)Hのように分極し、O–O結合のイオン的解裂によってFe=O+ -OHが生成する必要がある。CcPのヘム近傍に存在するHisのN-Hは、脱離する-OHと水素結合することで分極構造 (遷移状態) を安定化するが、MbのHisは両方の酸素原子とほぼ等距離にあり、その分極構造を安定化させる事は出来ない。そこで、MbのHis64の位置をペルオキシダーゼ類似の位置に移動することにした。具体的には、His64を立体障害の少ないロイシン (Leu) に置換し、29番目のアミノ酸であるLeu29あるいは43番目のフェニルアラニン (Phe43) をHisに置換したミュータントを作成した。それぞれのミュータントをH64L/L29H Mb、H64L/F43H Mbと呼ぶことにする。この他に、クロロペルオキシダーゼ(CPO)のヘム近傍を参考に、His64をグルタミン酸に置き換えた一連のH64D Mbミュータントを作成した。それらを用いた酸化反応の結果を表2に示す。高い不斉選択性と反応性の大幅な増加は明らかである。同様の高い不斉酸化反応は、オレフィン類のエポキシ化反応でも達成することが出来た。なお、過酸化水素とMbミュータントの反応で、O=Fe(IV)Por+•が生成していることは、ストップトフロー法による吸収スペクトルによって確認した6)。
表2 Mbミュータントによる不斉酸化
3.芳香環の選択的水酸化反応
我々の作成したMbミュータントが、高い不斉選択性と酸化活性を持っていることを紹介したが、芳香環やC–H結合の水酸化はほとんど進行しない。ヘム酵素の中で、こうした高難度酸化反応を行うのがシトクロムP450である。他のヘム酵素では達成できない水酸化反応をP450が効率よく進行させる理由は明確ではない。しかしながら、P450の特徴である軸配位子にシステイン由来のチオレートを用いているという点に注目している研究者は多い。これに対し、我々は少し異なる見解を持っている。例えば、図2(A)に示したP450camの結晶構造を見ると、基質であるd-カンファーがヘム鉄の真上に位置していることが分かる。このように、酸化活性種であるcompound Iが出来たときには、直ちに反応できる状態が準備されている。これに対して、ミオグロビンミュータントの場合には、遠位ヒスチジンをロイシンなどの小さなアミノ酸に置き換えた事によって基質を取り込める空間は準備されているものの、基質を保持するための機構は用意されていない。したがって、compound Iが生成したときに基質がヘム近傍にいるわけではない。そこで、基質をミューテーションによりヘム近傍に予め準備することを検討した。具体的には、ミオグロビンの43番目のアミノ酸であるフェニルアラニンをトリプトファンに置き換えた。同時に、compound Iを不安定化する要因である遠位ヒスチジンをロイシンに置き換えた (図2(B))。ロイシンを導入した理由は、遠位ヒスチジンをロイシンに置換したミオグロビンミュータントが、mCPBAを酸化剤に用いると定量的にcompound Iを与えることを既に明らかにしているからである。
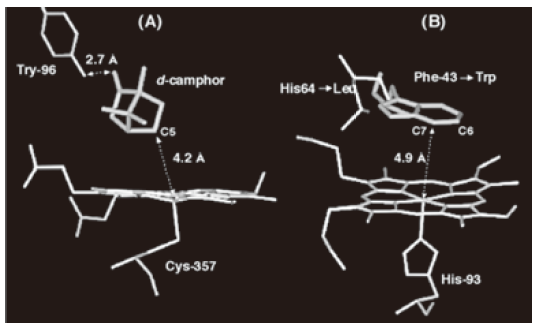
図2 P450camとF43W/H64L Mb のヘム近傍構造の比較
トリプトファンミュータントと酸化剤である過酸化水素の反応に伴う質量変化をESI Massにより測定すると、1当量の過酸化水素で反応はほぼ完結し、分子量は16Da増加する。どのアミノ酸が酸化修飾を受けているのか、この段階では明かではない。そこで、加水分解酵素によりフラグメントに分解し、液体クロマトによる溶出物の質量分析を行うと、43番目のトリプトファンを含む6残基のフラグメントが過酸化水素との反応により消失し、新たなピークを与えた。最終的には、このフラグメントを単離し、H-NMRによりインドール環の6位が選択的に水酸化されていることが明らかになった (図3)7)。
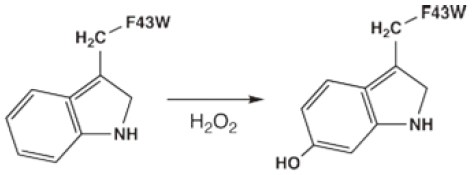
図3
ストップトフロー法による反応追跡の結果、この反応は2秒程度で完結することが分かり、ミオグロビンでも、基質が活性中心近傍に存在しさえすれば、ほぼ100%の効率で水酸化反応が進行することを明らかにした意義は大きいと考える。
このように、タンパク質の部位特異的な改変は、人工的な金属酵素を作り上げる上で非常に重要な手法であることが分かる8)。
4.合成錯体でミオグロビン活性中心を再構成
上記のような手法で作成したミオグロビンミュータントは、有機基質に対する一酸素添加反応 (monooxygenation) のみならず、過酸化水素の分解 (catalase反応) や基質の一電子酸化反応 (peroxidase反応) も触媒するなど、ヘム酵素全般の反応機構を検討・実証する良いモデルタンパク質であるという意味で、重要な結果だと自負している。しかしながら、このアプローチは、「すでに存在するヘム酵素の機能をミオグロビンに導入」したに過ぎず、天然には存在しない機能を賦与する事は出来ない。
そこで、ミオグロビンからヘムを取り除き (apo-Mbと呼ぶ)、様々な反応を触媒する金属錯体や有機金属化合物によってミオグロビンを再構成することにした。ヘムは、ミオグロビンの64番目のアミノ酸であるヒスチジン (His64) に配位しているので、合成錯体もHis64に配位することが出来る構造にすればよいと考えた。合成錯体で再構成されるミオグロビンの作成は、当初考えていたほどには簡単ではなかった。ヘムは400 nm付近にsoret帯と呼ばれる吸光係数の大きい特徴的な吸収があり、UV-visスペクトルで直ぐに見分けることが出来るが、非ヘム系の合成錯体には特徴的な吸収がないため、再構成が出来たか否かを簡便に判断できない。さらに、His64に錯体が配位したとしても、親和性が低い場合、単離・精製の段階で錯体がタンパク質から抜け出る可能性が高く、錯体配位子と周辺アミノ酸間の相互作用、例えば疎水相互作用やπ–π相互作用などを考慮した配位子設計が必要となる。
数年の泥コネを経て、図4に示すシッフ塩基錯体が、ヘムの位置に配位結合や疎水相互作用などによって安定に取り込まれていることが結晶構造から明らかになった。その後、図5に示すように、様々な配位構造の錯体や有機金属化合物をapo-Mbと再構成することに成功している9)。結晶構造からも明らかなように、再構成された人工金属タンパク質や有機金属タンパク質の金属周辺には、様々な反応を進めるための充分な空間がないために、期待された触媒反応活性は非常に低いものであった10)。
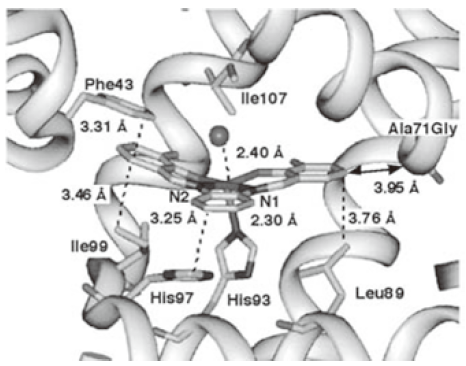
図4 Fe(Schiff base)•Mbの結晶構造
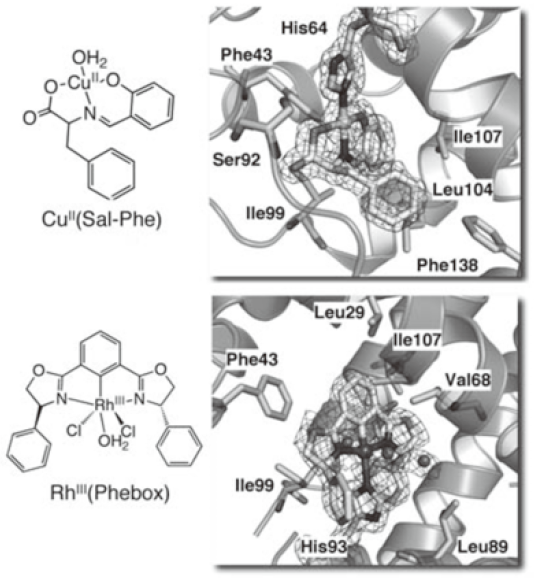
図5 CuやRh錯体を取り込んだMbの結晶構造
5.過酸化水素が使えるP450
野生型ミオグロビンとは異なり、はじめから高難度酸化反応が可能な高い活性を持っているP450は魅力的ではあるが、P450camを含めるほとんどのP450は、酸化活性種(O=Fe(IV) Por+•)の生成に還元剤として非常に高価なNADHもしくはNADPHを必要とするため、実験室スケールの反応でも気軽に利用できる酵素とは言えない。ペルオキシダーゼと同じように、P450の酸化活性種生成にも過酸化水素が使えれば、コストパフォーマンスに優れたバイオ酵素となるはずである。そこで我々は、P450の中でも非常に珍しく、過酸化水素を用いて長鎖脂肪酸を水酸化することができるP450BSβを利用しようと考えた11)。ところが、P450BSβは長鎖脂肪酸を高選択的に水酸化する (ある意味) 非常に優れた酵素であったため、他の基質の酸化反応にはまったく適用できないという弱点があった。P450BSβの高い基質特異性は、その独特の反応機構に基づくものである。長鎖脂肪酸 (パルミチン酸) は図6に示すように、そのカルボキシル基がヘムの近傍にある242番目のアルギニンと相互作用することによってヘム上方に固定化される12)。このカルボキシル基は、長鎖脂肪酸の固定化だけでなく、過酸化水素を用いた酸化活性種生成において、CPOのグルタミン酸と同じようにO-O結合のイオン的解裂を促進する (図7)。
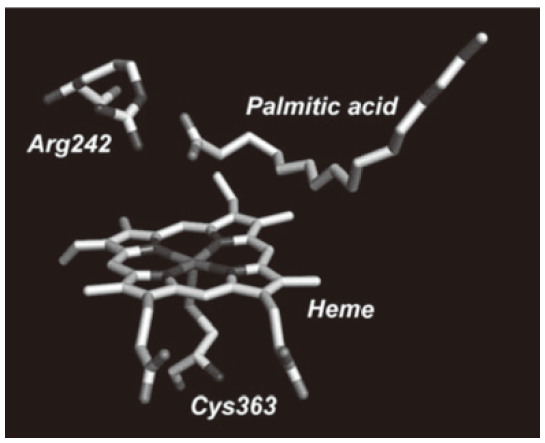
図6 基質結合型P450BSβのヘム近傍構造
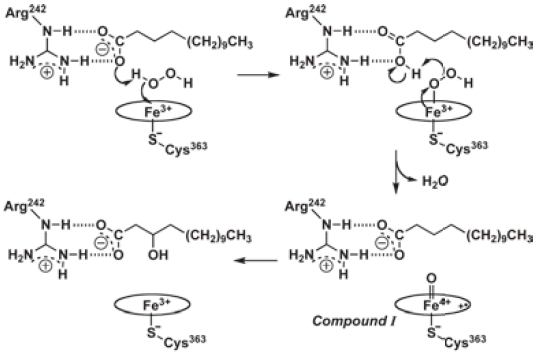
図7 P450BSβによるミリスチン酸の水酸化反応機構
長鎖脂肪酸のカルボキシル基が反応開始の引き金となっているので、長鎖アルカンやアルデヒド、アルコールなどカルボキシル基を持たない基質を酸化できない仕組みとなっている。P450BSβが酸化することができる対象基質は限られる。そこで我々は、「カルボキシル基を持ち、なおかつ、それ自体は水酸化されることのない擬似基質 (デコイ分子) 」をP450BSβに取り込ませることにより酸化活性種を生成させ、さらに添加した他の外来基質を酸化する反応システムを考案した (図8)。
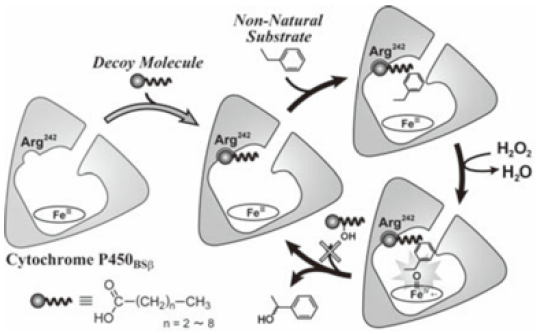
図8 基質誤認識システム
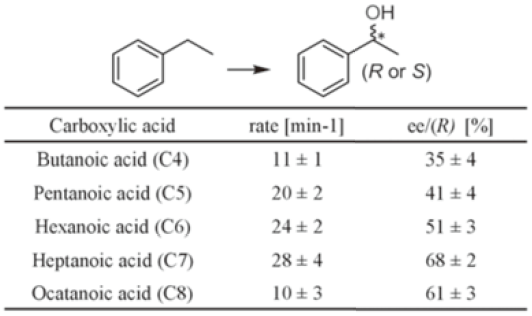
様々な分子を検討した結果、アルキル鎖長が短いアルキルカルボン酸が、P450BSβに水酸化されないデコイ分子として利用できることが分かった。そこで、炭素数7のヘプタン酸を反応系に添加して、グアイヤコールの一電子酸化反応 (ペルオキシダーゼ反応) を行ってみたところ、毎分3,000回転を越す速さで反応が進行した13)。デコイ分子を添加しない場合や本来の対象基質であるミリスチン酸を添加した場合には、ペルオキシダーゼ反応はほとんど進行しない。P450BSβによるミリスチン酸の水酸化反応活性は、毎分355回転と報告されており、本来の酸化活性を遥かに凌ぐ活性を示したことも興味深い。グアイヤコールの一電子酸化反応以外にも、スチレンのエポキシ化反応やエチルベンゼンの水酸化反応といった一酸素原子添加反応も進行した。エチルベンゼンの水酸化では、用いるデコイ分子のアルキル鎖長の違いにより酸化活性だけでなくエナンチオ選択性も大きく変化した (表3)。
表3 エチルベンゼンの水酸化反応
我々は、「デコイ分子がP450BSβに基質として誤認識され反応が進行する」反応系を基質誤認識システムと呼ぶことにした。基質誤認識システムは、部位特異的変異導入を必要とせず、野生型酵素をそのまま用いながら、その基質選択性を大きく変化させることができるこれまでに例をみないシステムである。ところで、「カルボキシル基を部位特異的変異導入により配置したら反応は進行するようになるのだろうか?」と誰もが知りたくなるような疑問が出てくる。そこで、242番目のアルギニンをグルタミン酸に置換したArg242Glu変異体を作成してみたが、まったく活性を示さなかった14)。ヘム周辺の他のアミノ酸をグルタミン酸に置換した変異体も作成したが、同様に酸化活性を示さなかった。したがって、現時点では、デコイ分子を用いる手法が、唯一のP450BSβの基質特異性変換法である。
ヘプタン酸を取り込んだP450のX線構造解析では、242番目のアルギニンの近傍にヘプタン酸のカルボキシル基に対応すると考えられる電子密度がみられ (図9)、ヘプタン酸のカルボキシル基が、長鎖脂肪酸のそれと同じようにO–O結合のイオン的解裂を促進していると考えられる。また、ヘプタン酸のアルキル鎖に対応する電子密度が見られないことから、アルキル鎖部分は固定化されずに揺らいでいることが示唆された。このことは、ヘプタン酸が水酸化反応を受けない事実を支持している15)。
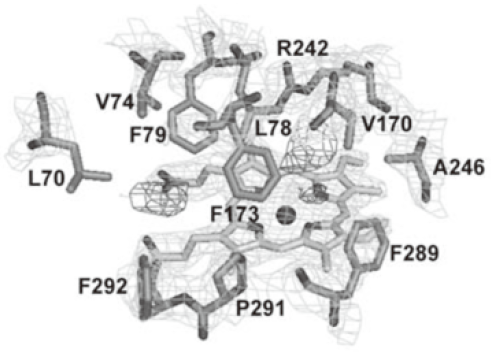
図9 P450BSβ-ヘプタン酸共結晶のヘム近傍構造
最近、基質誤認識システムにより、芳香環の水酸化反応も進行することが明らかとなり、1-メトキシナフタレンの水酸化反応が脱メチル化反応に優先して起こることがわかっている。基質誤認識システムは変異導入と組み合わせることで、さらに幅広い基質の酸化に適用できると期待している16)。
6.おわりに
本稿では、最近我々が行っている次世代に繋がる人工金属酵素の創製のための試みを紹介してきた。ミオグロビンタンパク質の内部空間を利用する人工金属タンパク質の設計や、P450の反応を外部から添加する物質により制御する研究は、従来の研究とは本質的に異なる独創的な研究であると自負するものであるが、まだまだ努力不足であることは率直に認めざるを得ない。今後、有機化学や錯体化学分野の研究者が、タンパク質をあたかも試薬の一つであるかのように扱う時代が来ることを夢見て、本稿を閉じたい。研究室の諸氏には深く感謝するものである。
文献
1) 赤井周司, 北泰行: 有機合成化学協会誌, 65, 772 (2007).
2) 渡辺芳人: タンパク質・核酸・酵素, 49, 22 (2004).
3) Watanabe, Y.: In Model studies on heme monooxygenases , Funabiki, T. (ed), Oxygenases and Model Systems, Kluwer Academic Publishers, Netherlands, 19, 22 (1997).
4) (a) Watanabe, Y., Numata, T., Iyanagi, T., Oae, S.: Bull. Chem. Soc. Jpn., 54, 116 (1981). (b) Watanabe, Y., Oae, S., Iyanagi, T.: Bull. Chem. Soc. Jpn., 55, 188 (1982).
5) Groves, J. T., Watanabe, Y.: J. Am. Chem. Soc., 110, 844 (1988).
6) Ozaki, S., Roach, M. P., Matsui, T., Watanabe, Y.: Acc. Chem. Res., 34, 818 (2001).
7) Pfister, T. D., Ohki, T., Ueno, T., Hara, I., Adachi, S., Makino, Y., Ueyama, N., Lu, Y., Watanabe, Y.: J. Biol. Chem., 280, 12858 (2005).
8) Watanabe, Y., Nakajima, H., Ueno, T.: Acc. Chem. Res., 40, 554 (2007).
9) (a) Ohashi, M., Koshiyama, T., Ueno, T., Yanase, M., Fujii, H., Watanabe, Y.: Angew. Chem. Int. Ed., 42, 1005 (2003). (b) Ueno, T., Ohashi, M., Kono, M., Kondo, K., Suzuki, A., Yamane, T., Watanabe, Y.: Inorg. Chem., 43, 2852 (2004). (c) Ueno, T., Koshiyama, T., Ohashi, T., Kondo, T., Kono, M., Suzuki, A., Yamase, T., Watanabe, Y.: J. Am. Chem. Soc., 127, 6556 (2005).
10) (a) Abe, S., Ueno, T., Reddy, P. A. N., Okazaki, S., Hikage, T., Suzuki, A., Yamane, T., Nakajima, H., Watanabe, Y.: Inorg. Chem., 46, 5137 (2007). (b) Satake, Y., Abe, S., Okazaki, S., Ban, N., Hikage, T., Ueno, T., Nakajima, H., Suzuki, A., Yamane, T., Nishiyama, H., Watanabe, Y.: Organometallics, 26, 4904 (2007).
11) Matsunaga, I., Ueda, A., Sumimoto, T., Ichihara, K., Ayata, M., Ogura, H.: Arch. Biochem. Biophys., 394, 45 (2001).
12) Lee, D.-S., Yamada, A., Sugimoto, H., Matsunaga, I., Ogura, H., Ichihara, K., Adachi, S., Park, S.-Y., Shiro, Y.: J. Biol. Chem., 278, 9761 (2003).
13) Shoji, O., Fujishiro, T., Nakajima, H., Kim, M., Nagano, S., Shiro, Y., Watanabe, Y.: Angew. Chem., Int. Ed., 46, 3656 (2007).
14) Shoji, O., Fujishiro, T., Nagano, S., Shiro, Y., Watanabe, Y.: unpublished results.
15) Shoji, O., Fujishiro, T., Nagano, S., Shiro, Y.: Watanabe, Y.: manuscript in preparation.
16) Shoji, O., Wiese, C., Fujishiro, T., Bernhard, W., Watanabe, Y.: manuscript in preparation.
![]()