【トピックス】
動物細胞を用いたタンパク質生産 ―そのポテンシャルは如何ほどか―
大政健史
徳島大院・ソシオテクノサイエンス研
1.はじめに
酵素工学と動物細胞とは関連があまりないように感じられる会員の方も多いのではと思われる。一方、ここ10年の間に動物細胞を用いたタンパク質生産は飛躍的にその濃度が上昇し、抗体ならば10 g/Lの生産例も報告されている。さらに、意外に思われるかも知れないが、動物細胞を用いた「酵素」生産は医薬品分野においては、年間数百億円の売り上げがあり、さらに近年では、バイオ後続薬 (バイオジェネリック、バイオシミラー、フォローオンバイオロジックス) の生産系としても注目されている。また、抗体に限ると、その培養にかかるコストがg蛋白質あたり数ドル程度にまで低下しており、非常に安価なタンパク質生産が可能となっている。
本稿ではタンパク生産系としての動物細胞の現況とポテンシャルを紹介するとともに、その問題点についても明確にしたい。これをきっかけにして、動物細胞系にも興味をお持ち頂き、是非多数の方々にご利用頂けると幸いである。課題の性質上、雑駁な内容になることをお許し願いたい。
2.歴史的経緯と現状
生体外動物細胞培養は、今からほぼ100年前に始まり、当初は実験室内における細胞生物学的な解析に用いられた。生体外培養技術は職人的なノウハウが必要とされ、一般的に用いられにくい実験手法であったが、この30年ほどの間に、培養方法、培地、さらには細胞株の改良など飛躍的な進歩があり、原理さえ踏まえていれば誰でも用いることの可能な「工学」へと変貌を遂げている。これに伴って、動物細胞の産業利用も飛躍的に発展し、数万Lにわたる大規模培養や、年間何千億円もの売り上げがあるタンパク質医薬品生産、次世代の産業を担うと期待されているES細胞 (胚性幹細胞)、iPS細胞 (誘導多能性幹細胞) などに代表されるティッシュエンジニアリング分野まで、幅広く用いられるようになった。バイオベンチャー企業として有名なGenentechやAmgenが、現在は動物細胞を用いたタンパク質医薬品が主力の世界有数の製薬企業へと成長している点からも、この分野の発展を感じ取ることができる。
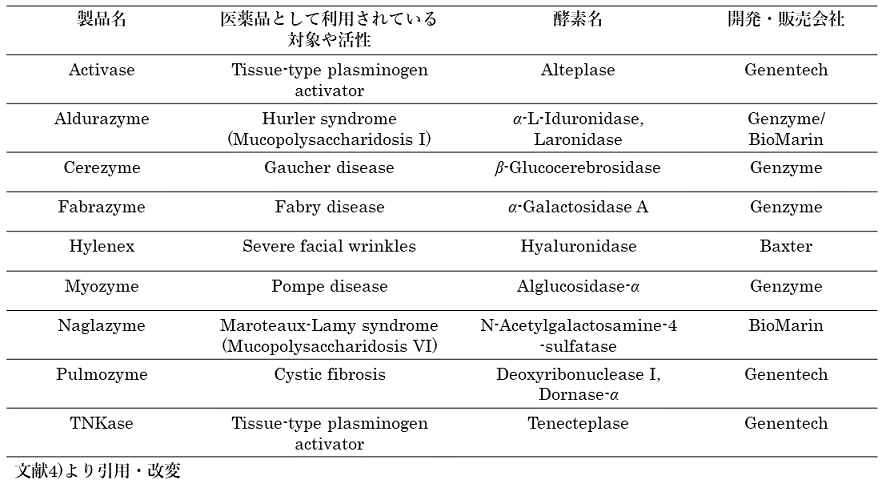
現在、タンパク質医薬品生産場としての動物細胞とは、チャイニーズハムスター卵巣 (CHO) 細胞が主流である。2006年にアメリカとEUにおいて実際に上市されているタンパク質医薬品107品目 (抗体医薬29品目を除く) のうち、大腸菌 (Escherichia coli) が42%、CHO細胞が29%、酵母 (Saccharomyces cerevisiae) が21%、すなわちこの3つの宿主細胞で実に90%以上のタンパク質医薬品が生産されており1,2)、近年の技術開発もCHO細胞を中心に行われている。タンパク質医薬品の大部分は成長因子や抗体などであるが、タンパク質医薬品としての組換え酵素もCHO細胞を用いて生産されている。例えば、2006年に米国においてはタンパク質医薬品としての治療用酵素だけでも、0.76Billion $ (750億円) の売り上げがあり3)、Cerezyme (β-Glucocerebrosidase)、Pulmozyme (Deoxyribonuclease I, Dornase-α)、Fabrazyme (α-Galactosidase A) のCHO細胞で生産される3品目で売上げ額の70%以上を占めている。表1に欧米においてCHO細胞を用いて生産される組換え酵素医薬品についてまとめて示す。これらの酵素医薬品は近年、バイオ後続薬の開発対象として注目されており、これらの後続薬の生産においても、CHO細胞が選択される場合が多い。
表1 欧米において上市されている組換えCHO動物細胞を用いて生産される酵素医薬品4)
3.動物細胞を用いたタンパク質生産の特徴
タンパク質生産として動物細胞を用いる際の特徴として、(1) 適切な分泌シグナルを付加することにより、通常細胞外に分泌される、(2) 微生物や下等真核生物、昆虫細胞などと比較して翻訳後修飾プロセスが発達しており、複雑な構造や翻訳後修飾を持つ高等真核生物 (特にヒト) 由来の生理活性物質の生産がほとんど問題なく可能、(3) 細胞増殖が微生物より遅く、発現株構築に時間がかかり、さらに微生物ほどの高密度培養が達成しがたい。(4) 微生物による生産と比較して生産性が低く、非常に高コストと一般的に言われている、などが挙げられる。以下では、いくつかの点について解説する。
(1) 宿主-ベクター系と細胞株構築5)
動物細胞に於ける発現系構築は、現在は市販ベクターに目的遺伝子を組み込むことでほとんどのタンパク質の発現系構築が可能である。一方では、高レベルのタンパク質生産系として構築する場合には、単に遺伝子を組み込むだけではなくて、その後の遺伝子増幅に代表されるgene dosage効果の実践、ならびに高生産株構築のための細胞選抜などのステップが必要である。
動物細胞に於ける発現系は染色体外において自律複製可能なプラスミドDNAがほとんど無いため、非常に組み込み確率の低い染色体内への相同組換えを用いて発現系が構築される。発現ベクターは通常、陽性に荷電したリポソームとDNAの複合体を形成させ、細胞の自発的な取り込みによって遺伝子導入を行うリポフェクション法を用いて細胞に取り込ませる。染色体に組み込まれた細胞をベクターに存在する選択マーカーにより選択することで、恒常発現株が構築可能となる。
発現系構築におけるCHO細胞の特徴は、恒常発現株作成の容易さと選択された細胞株の多様性にある。同じベクターを同じ宿主CHO細胞に導入して得られた発現株は通常同じ生産性を持つのではなく、非常に幅広い多様性を持つ。これは、染色体上のエピジェネテッィクな環境により、発現が影響されるためと考えられている。また、CHO細胞特有の半接合状態の染色体もこの幅広い多様性に関連すると思われる。いずれにせよ、よりたくさんの細胞株をスクリーニングすることにより、より生産性の高い細胞株を構築することが可能となる。そのため、ロボットを用いたスクリーニング系も利用される。一方、これらのスクリーニングの労力を軽減するために、産生株の取得効率を上昇させる工夫もなされている6)。
(2) 分子多様性と翻訳後修飾
生産されたタンパク質の分子多様性については、実験室レベルではあまり留意される事がないように思われる。一方、大規模タンパク質医薬品生産の現場では、この分子多様性は常に意識されている問題である。これは、動物細胞を用いて生産させるタンパク質は、糖タンパク質が多く、複雑な構造や翻訳後修飾されるため、様々な不均一性が生じている。Genentech社のHarrisはCHO細胞を用いた遺伝子組換え抗体の商業生産における分子多様性について、(1) 抗体Fc鎖における糖鎖修飾の多様性、(2) H鎖におけるリジンのプロセッシング、(3) メチオニン残基の酸化、(4) ヒンジ領域のフラグメント化、(5) 糖付加、(6) アスパラギン残基の脱アミノ化およびアスパラギン酸残基の異性化、(7) システイン残基におけるSS結合解除、等の例を報告している7)。
この中でも、特に糖鎖修飾の多様性は、糖鎖が生産されたタンパク質の生体内機能に直接関連するため注目されている。CHO細胞において生産された糖タンパク質における糖鎖修飾は、ほぼヒトに近い構造の糖鎖が付加するものの分布が存在し、宿主細胞の生産性、培養条件によって糖鎖修飾の多様性が変動することが知られている8)。この多様性変動は、糖タンパク質分泌において、糖鎖修飾が、タンパク質のポリペプチド鎖が合成された後に付加、刈り込み、さらに転移反応などの複雑な酵素反応ステップによって糖鎖修飾が行われるために起こる。実際の生産においては、培養条件による糖鎖構造の変動も考慮にいれた生産系構築が行われている。
(3) 現在の細胞培養法とコスト・生産性比較
動物細胞大量培養を用いたタンパク質生産はリンパ芽球によるインターフェロンの生産 (8,000 L) に代表される様に、1970年代から既に実用化されている。通常、リンパ球由来細胞を除き、動物細胞は増殖に足場を必要とする。この時点での細胞培養は固定化酵素と同様の手法、すなわち、固定化担体としてのマイクロキャリア (細胞の接着可能な微小なビーズ) 上に細胞を自然に接着進展させ、キャリア表面において増殖させることにより、細胞培養を行っている。現在でも、本手法は細胞培養を用いたワクチン生産において活用されている。
技術的な観点からみると、マイクロキャリアを浮遊して培養させるということは、微生物による発酵槽を用いた深部培養による物質生産と原理的にはほぼ同じ手法となる。さらに近年の長足な進歩は無血清浮遊化培養法にある。1970、80年代に実用化されていた細胞培養には遺伝子組換えは用いられていなかったが、90年代に入ると遺伝子組換えを施した細胞を用いてタンパク質を生産するようになった。その宿主として汎用されたのがCHO細胞となる。さらにCHO細胞の特徴は前にも述べたが遺伝子組換え細胞株構築の容易さにある。1990年代初頭まで牛胎児血清を用いる血清培地にて生産系が構築されていたが、近年は牛海綿状脳症 (BSE) を始めとする人獣共通感染症への懸念から無血清培地が必ず用いられる。特にCHO細胞は血清培地から無血清培地に馴化させやすく、様々な無血清培地も市販されていることから、無血清培地を用いて培養を行うことが比較的容易な細胞である。無血清に馴化することにより、リンパ球系細胞と同様に浮遊撹拌培養が可能となり、マイクロキャリアに代表される接着担体も不要となり、コストの削減およびスケールアップの容易さが実現できるようになった。
また、スケールアップ時における動物細胞培養特有の問題点も解決されるようになっている。動物細胞は撹拌時に生じるせん断力に弱く、撹拌回転数をあまり上昇させることができない。また、無血清培地とはいえ、培地中にタンパク質成分を含むため泡が生じやすく、泡による細胞へのダメージがあり、通気量をあまり上昇させることができない。そのため問題となるのが培地中の溶存炭酸ガスの蓄積である。エネルギー源 (グルコース、グルタミン) の代謝によって生じた溶存炭酸ガスが抜けにくくなり、通常の通気による酸素供給だけでなく、溶存炭酸ガスの脱離についても考慮したスケールアップも行われる必要がある9)。
タンパク質医薬品の中でも抗体はその人体への投与量の多さから、産業レベルでの実生産において成長因子やサイトカインに比べても大量生産が要求されている。ここ10年、特にCHO細胞における抗体生産技術は飛躍的に向上し、近年では最大10 g/Lのレベルの抗体生産がいくつか報告されている10)。では、g/Lレベルでの生産を可能にしている因子は何であろうか。もちろん高生産株の選抜、構築も貢献しているが、特に、飛躍的な生産性向上に貢献しているのは培養方法の検討である。細胞培養における培養操作は微生物培養の場合と同様に、回分、流加、連続の3種類に分類できる。特に80年代後半から90年代前半は、培地を連続して供給し、老廃物を含んだ培地を連続的に引き抜くという連続灌流培養法が盛んに開発された。
連続灌流培養法は細胞を固定化 (血清培地の場合は上述のように自然に接着するため、固定化操作が不要) もしくは細胞分離装置 (細胞と培地の比重差を用いて分離) を用いることにより、細胞を系内に高濃度に保ったままで連続高生産が実現できるため、より小規模の設備で、大量培養と同様の生産性を上げることが可能となる。また、生産された生産物が不安定で分解しやすい場合にも、生産物を直ちに回収できる利点があった。ただし、連続灌流においては、雑菌汚染無く安定して長期間 (少なくとも数ケ月以上) 運転する必要があり、その特殊性と、必ずしも使用培地量が軽減されないことから、応用例はRemicade (Centocor社1998)、Cerezyme (Genzyme社1994)、Gonal-F (Serono社1997)、Kogenate-FS (Bayer社2000) 等、いくつかに限定されている11)。近年、いくつかの新しい灌流装置が考案されているが、この問題点を積極的に解決するには至っていない。
現在、工業的な大量培養において最も用いられているのは微生物でも主流の流加培養である。通常の動物細胞の場合、細胞の増殖と共に培地中の栄養源が減少し、乳酸、アンモニアなどの有害代謝産物が蓄積するため、細胞増殖期間を長く保つことができない。また、細胞壁が無いため、浸透圧変化に弱く、最初から高濃度の基質を添加して培養を行うことも困難である。そこで、高濃度の栄養源・成長因子を含む培地を流加することにより、栄養源・成長因子の供給、有害代謝産物の希釈を行い、培養時間を延長させ、高細胞濃度を達成し、高タンパク質生産を実現する流加培養法が培養操作の中心として用いられている。さらに最近特に利用が盛んになっているのは使い捨て (ディスポーザブル、シングルユース) のリアクターである。医薬品生産にはGMP基準を満たすために、バリデーションと呼ばれる洗浄や滅菌操作、リアクターの安定運転にかかる検証が必要である。すなわち実際のリアクターを運転する時間よりも、これらの検証操作にかかる時間が非常に長期にわたり、そのメンテナンスコストが問題となってきた。ディスポーザブルリアクターを用いると、既に検証の終わったリアクターが納品され、さらにリアクターの蒸気滅菌等の操作が不要となるため、エネルギーコストも軽減される。現在、GMP基準を満たす撹拌翼もついた1,000 Lレベルのディスポーザブルリアクターも既に多数市販されている。また、ガンマ線滅菌を行うため、センサー類も使い捨ての蛍光プローブを用いることが可能となり、蒸気滅菌の制限のある微生物よりもさらに高度な培養が可能になると考えられる。
では、実際には微生物でのタンパク質合成と比較して、動物細胞はどの程度の生産性・コストを持っているのであろうか。直接比較可能なパラメータとして比生産速度 (細胞あたり、単位時間あたりの生産量) がある。高生産を実現可能なCHO細胞の場合、抗体比生産速度20~80 pcd (pg-1 cell-1 day-1) 程度の値が報告されている。CHO細胞の正確な乾燥細胞重量はデータがほとんどないが、ハイブリドーマ細胞で得られている値を参考に、乾燥細胞重量を300 pg/cellと仮定すると、抗体比生産速度は2.7~11.1 (mg-protein-1 g-dry-cell-1 h-1) 程度と推定換算される。同じく10 g/Lレベルの高生産が報告されているメタノール資化性酵母Pichia pastorisの流加培養によるヒトアルブミン生産の比生産速度0.1~0.3 (mg-protein-1 g-dry-cell-1 h-1)12) と比較すると、CHO細胞自体の生産性は相当高いレベルにあると推定される。
動物細胞によるタンパク質合成は、微生物や他の発現系による生産と比較して、非常に高コストと一般的には言われている。組換え植物によるタンパク質生産の優位さを論じた論文においては、1 gのタンパク質を生産するための費用が微生物では1ドル、植物では0.1ドル、CHO細胞では300ドル必要と述べている例もある (試算の根拠は不明)13)。では、実際のコストはどの程度であろうか。古い例になるが、実際に医薬品生産として用いられているティッシュプラスミノーゲンアクティベーター (tPA) 生産を大腸菌と直接比較した例では、なんと大腸菌に比べて半分程度の試算になっている14)。さらに近年では、10トンタンクにて5 g/Lの最終濃度でCHO細胞を用いて無血清培地を用いてGMPレベルにて流加培養生産した場合、1 gのタンパク質を生産するのに必要なコストは26ドルであり、そのうち、培養コストは2ドル、精製に4ドル、その他は医薬品として出荷に必要なコストや労働コストであるという試算もある15)。この数値はあくまでもモデルケースであるが、現在の無血清培地を用いたCHO細胞のg/Lレベルの高生産流加培養でのコストは、微生物と比較してもさほど遜色ないレベルであると考えられる。さらに、微生物等の発現系に比較してCHO細胞培養系の場合は、培地中に分泌される生産物以外の混在が少なく、生産物の分解もあまりされず、したがって精製が比較的簡便に行えるという利点があり、産業レベルでの生産に関しては、あえて他の生産系に置き換える必要性はもはや無いと言えよう。
4.今後の展望 ―いったい何が問題点なのか?
タンパク質合成の場として動物細胞、特にCHO細胞を用いる技術は、本稿に紹介した様に近年飛躍的に向上している。また、その生産性も微生物に比較しても高く、さらに分泌生産が可能なため、精製にかかるコストも比較的抑えられる。では、一体何が問題点であろうか。
大きな問題点は、(1) 同じベクターをおなじCHO細胞に導入して組換え細胞を構築しても、その生産性はバラバラである、(2) 得られた細胞株を「育種」、すなわち選抜したり、遺伝子増幅などの手段を用いないと高生産株が得られない、(3) 構築した細胞株の最適な培養法も株ごとにバラバラである点にある。言い換えれば経験的な手法と大量スクリーニングにより高生産系を構築できるものの、その中身はブラックボックスのままになっている。
我々のグループでは、その一端を解明するために、CHO細胞のゲノム解析を進めている16)。意外に思われるかもしれないが、CHO細胞は、国内の売り上げだけで何千億円もある製品を生み出している宿主細胞でありながらそのゲノムは全く解析されていない。この原因ははっきりとはわからないが、(1) 元々のチャイニーズハムスターが実験動物として用いられてないため、モデル生物として基礎生物学や基礎医学から要望が無かった、(2) CHO細胞のゲノムはチャイニーズハムスターからかなり変動しており、サイズの大きさ (ヒトより少し小さい程度の2.8 Gと推定される) とあいまって、コストもかかり解析が難しい、が理由として考えられる。現在、CHO細胞の全ゲノムの5倍長をカバーするバクテリア人工染色体 (BAC) ライブラリーを構築し、CHO細胞の染色体変化について解析している。
また、動物細胞特有の翻訳後プロセスについても解析/制御する必要がある。我々のグループでは、小胞体ストレス応答に関わる因子を強化したCHO細胞を構築することにより、生産性を約2倍に高める事や17)、ゴルジ体内へのフコースの輸送を抑えることによるフコース修飾低減にも成功しており18)、このような翻訳後修飾のプロセスを強化・制御するアプローチは、今後求められていると言えよう。また、現在のところCHO細胞は医薬品のGMPによる実生産に用いられる用途だけであるが、さらに改良が進めば医薬品以外の目的のタンパク質合成に対しても貢献することが可能と考えられる。

図1 CHO細胞の染色体電子顕微鏡写真
文献
1) 大政健史:化学と生物, 45, 9 (2007).
2) 大政健史:化学と生物, 48, 255 (2010).
3) Aggarwal, S.: Nature Biotechnol., 26, 1227 (2008).
4) 金 昱東、大政健史:「酵素利用技術体系」, エヌ・ティー・エス, 181 (2010).
5) 高木 睦、大政健史:「第5版 実験化学講座29巻 バイオテクノロジーの基本技術」, 丸善, 61 (2006).
6) Omasa, T., Onitsuka, M., Kim, W.-D.: Current Pharmaceutical Biotechnol., 11, 233 (2010).
7) Harris, R. J.: Dev. Biol. (Basel) , 122, 117 (2005).
8) 大政健史、菅健一:「動物細胞工学ハンドブック」, 朝倉書店, 198 (2000).
9) 松永直樹:「抗体医薬のための細胞構築と培養技術」, シーエムシー出版, 188 (2010).
10) 大政健史:日本生物工学会誌, 86, 393 (2008).
11) Kompala, D. S., Ozturk, S. S.: Cell culture technology for pharmaceutical and cell-based therapies, CRC press, 387 (2006).
12) Kobayashi, K., Kuwae, S., Ohya, T., Ohda, T., Ohyama, M.: J. Biosci. Bioeng., 90, 280 (2000).
13) Hood, E. E., Woodard, S. L. Horn, M. E.: Current Opinion in Biotechnology, 13, 630 (2002).
14) Datar, R. V., Cartwright, T., Rosen, C.-G.: Bio/Technology, 11, 349 (1993).
15) Kelley, B.: Biotech. Progress, 23, 995 (2007).
16) Omasa, T., Cao, Y., Park, J. Y., Takagi, Y., Kimura, S., Yano, H., Honda, K., Asakawa, S., Shimizu, N., Ohtake, H.: Biotech. Bioeng., 104, 986 (2009).
17) Ohya, T., Hayashi, T., Kiyama, E., Nishii, H., Miki, H., Kobayashi, K., Honda, K., Omasa, T., Ohtake, H.: Biotech. Bioeng., 100, 317 (2008).
18) Omasa, T., Tanaka, R., Doi, T., Ando, M., Kitamoto, Y., Honda, K., Kishimoto, M. Ohtake, H.: J. Biosci. Bioeng., 106, 168 (2008).
![]()