【トピックス】
酵素活性部位における「低障壁水素結合」
石北 央、斉藤圭亮
京大生命科学系キャリアパス形成ユニット/科学技術振興機構さきがけ、京大生命科学系キャリアパス形成ユニット
1.はじめに
酵素の活性部位では、反応における遷移状態 (transition state) を安定化することにより、活性化エネルギーを下げる。そのため、水溶液中では大変遅い反応でも、酵素中では速やかに起こり得る。遷移状態安定化には、特に水素結合による相互作用が重要な役割を果たす。水素結合のエネルギーは一般に1~4 kcal/mol程度である1)。一方、「低障壁水素結合 (low barrier hydrogen bond, LBHB) 」という特殊な水素結合のエネルギーは、20 kcal/molにも及ぶと言われている2,3)。そのようなLBHBが活性部位に存在すれば遷移状態の安定・反応の促進に著しく寄与するに違いない。また、新たな設計原理による創薬、酵素の機能改変としても参考になり得る。しかし一方で、「LBHB」の概念や定義はしばしば曖昧であった1,4)。酵素活性部位でのLBHBの存在を論じたCleland2) やFrey3) の論文がScience誌に発表された後は、蛋白質内の短い水素結合に対し慎重な検証もなく安易にLBHBと結論づける風潮も見られた。ここでは、蛋白質におけるLBHBの特徴を改めて整理してみたい。
2.低障壁水素結合の特徴
蛋白質内におけるLBHBの特徴に関しては、結晶構造解析、プロトン核磁気共鳴 (1H-NMR)、理論化学等を中心とした研究手法の立場からそれぞれの主張がなされてきた。
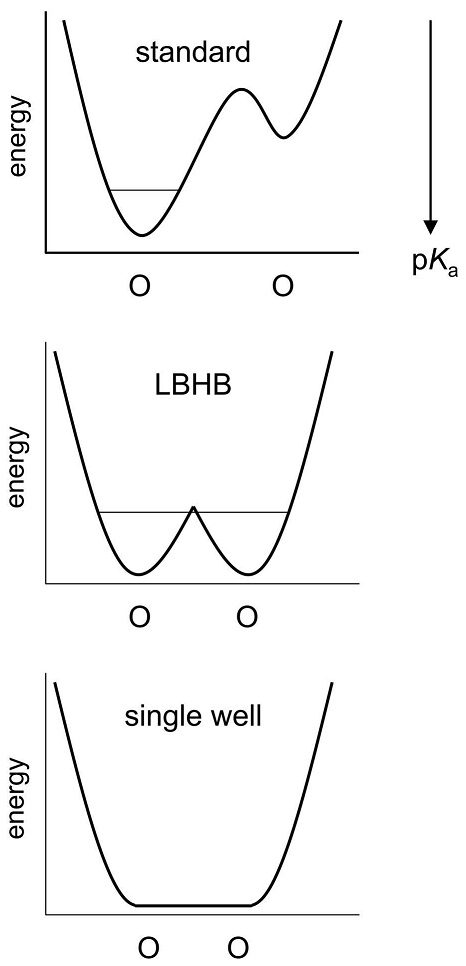
(NMRケミカルシフト (δH) と水素結合長) 「強い水素結合」においては、H原子はアクセプター側のO原子に引かれる傾向にあるため、電子の遮蔽効果が少なく、NMRでは低磁場側にシフトし大きなδH値を示す。Jeffrey5) やFrey6) によると、水素結合はδH値やO–O結合長のサイズにより以下のように分類できる。①ionic (single-well) H bond (δH=20–22 ppm、O–O結合長=2.4–2.5 Å)、②LBHB (δH=17–19 ppm、O–O結合長=2.5–2.6 Å)、③weak H bond (δH=10–12 ppm、O–O結合長 > 2.6 Å)。O–O結合長は、ionic H bondが最も短く、LBHBはそこまでは短くない (図1)。
(pKa値) Warshelによれば、結合長だけによるLBHBの判断は適切でなく、水素結合のポテンシャルエネルギー曲線が左右対称であることが必要である4) (図1)。ポテンシャルエネルギー曲線は、蛋白質環境内でのドナー・アクセプターのpKa値を知ることができれば、ある程度予測がつく。通常の水素結合 (=asymmetric double well potential) では、ドナー側のpKa値が、アクセプター側のpKa値より高い (=H原子がドナー側に引きつけられている)。なお、pKaの大・小関係は、ポテンシャルエネルギーの低・高に対応する (図1)。一方、ionic (single-well) H bondやLBHB形成においては、ドナー側とアクセプター側のpKa値がほぼ一致することが必須である。この条件は、Cleland2) やFrey3) の論文にも明記されており、多くの研究者が同意する特徴である。

(共有結合性) 通常の水素結合はHδ+とOδ–のような分極をもつ結合である。一方、LBHBでは、H原子が二つのO原子間を行き来できるため、電荷がドナーからアクセプター領域において非局在化して共有結合性をもつ2,3)。しかし、「LBHBの共有結合性」に関しては、これを唱え始めたClelandらの主張2) を慎重に検証する必要がある。Clelandによると、蛋白質内部は「無極性溶媒」のようなものであるため、電荷が非局在化したLBHBは容易に形成され得る、とのことである。しかし、「疎水的環境=無極性溶媒」は正しい理解ではない。
蛋白質内部には、蛋白質構成原子のvan der Waals半径 (体積) により、水分子は入り込みにくい。その結果、蛋白質内部では、水分子によるsolvation energyが得られにくい。これこそが「蛋白質内部の疎水性」である。一方、蛋白質内部には無極性溶媒とは異なり、多くのdipoleが存在する。極性アミノ酸側鎖だけでなく、主鎖カルボニル基Cδ+=Oδ–は強く分極しており、蛋白質内での電荷の安定に大きく貢献する7,8)。さらに、蛋白質中での静電相互作用は、バルク水中のように遮蔽されず、強い。このような場では、分極している通常の水素結合なら大きな安定化効果を得られるが、電荷が非局在化しているLBHBでは小さい。なお、蛋白質内部にある塩基性残基・酸性残基は、必ずしも電荷中性状態である必要もない。安定に電荷を持つか否かは、周辺にpolarな環境を持つかどうかであることを付記しておく。
3.Photoactive yellow proteinの活性部位での「LBHB」
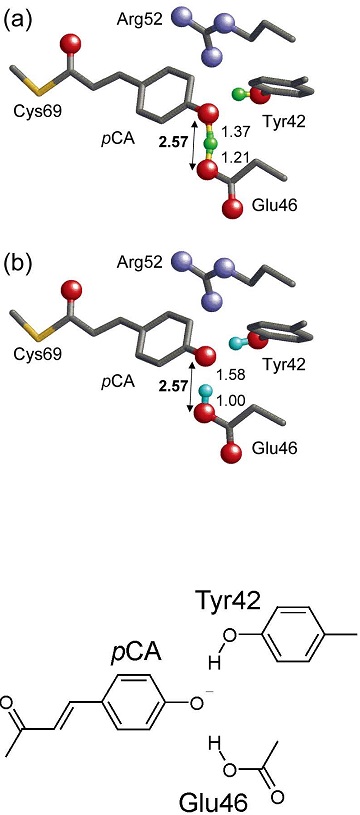
Photoactive yellow protein (PYP) の蛋白質の活性中心部位 (クロモフォア) は、主としてTyr42、Glu46、p-coumaric acid (pCA) からなり、Tyr42とpCA、Glu46とpCAがそれぞれ短い水素結合を形成している (OTyr42–OpCA=2.49–2.51 Å、OGlu46–OpCA=2.54–2.61 Å9))。過去にアメリカのグループが中性子構造解析 (2007年) を行ったが、分解能が2.5 Å程度でありこれらの水素結合がLBHBかどうかの判断は下せなかった10)。したがって、分解能向上は急務であった。2009年、中性子構造解析 (分解能1.5 Å) により、水素結合OGlu46–OpCAはLBHBであると報告された11)。その判断の根拠は以下の通りである。
(根拠1) 中性子構造解析の結果、H原子がOGlu46–OpCA (2.57 Å) においてほぼ真ん中の位置に存在していた (OGlu46から1.21 Å、OpCAから1.37 Å) 11) (図2)。
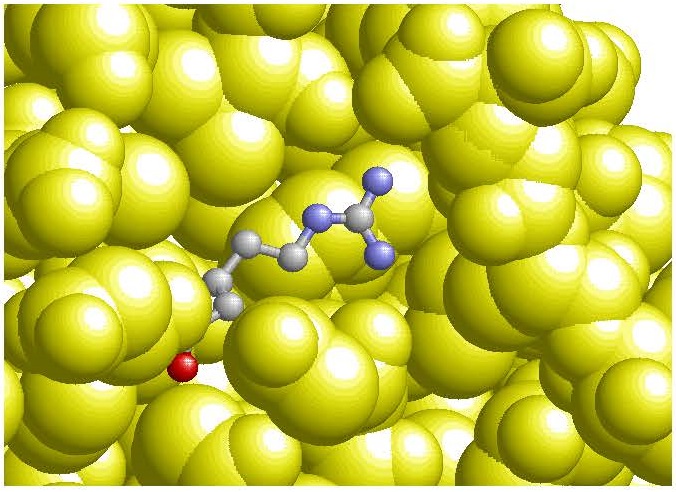
(根拠2) クロモフォアに最も近い塩基性残基Arg52がdeprotonateしていた11)。Arg52はX線結晶構造解析から、蛋白質表面に露出していることが既知であり (図3)、protonateし正電荷を持つ、と当然考えられてきた12)。ところが、今回の中性子構造解析ではArg52の側鎖のH原子が1個見えず、deprotonateしていると結論づけられた11)。
なぜdeprotonated Arg52がOGlu46–OpCAのLBHBを支持する根拠となり得るのか?クロモフォアTyr42、Glu46、pCAには負電荷 (-1) が存在する13-16)(図2)。したがって、「もし、Arg52が正電荷を持たなければ、蛋白質内部のクロモフォアの負電荷はエネルギー的に不安定なはずである。しかしGlu46とpCAがLBHBを形成して共有結合性を帯びていれば、Arg52の正電荷がなくても、負電荷は非局在化し安定に存在し得る。」と解釈され、deprotonated Arg52はLBHB存在の根拠として主張された11)。
![]()
![]()
4.Photoactive yellow proteinにおける「LBHB」の検証
これらの中性子結晶解析に基づく「解釈」11) (注;実験結果そのものではなく、あくまで結果の解釈) に対し、私たちは、quantum mechanical/molecular mechanical (QM/MM) 計算とelectrostatic titration計算を行った17)。 (なお、理論化学といっても、そこから得られる値は結晶構造の原子座標に基づいているため、あくまで結晶構造を忠実に反映した値である。例えばAsn側鎖のNとOの帰属が結晶ごとに異なる場合は、得られる酸化還元電位、pKa値等は当然異なる。逆に、そこから現実に即した側鎖の向きも推定できる18)。)
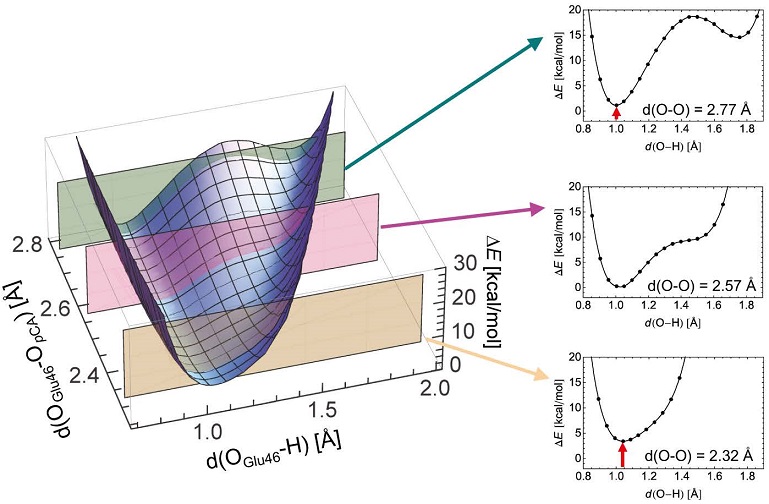
(水素原子位置) QM/MM計算では、OGlu46–OpCA結合距離が2.57 Åとなり、中性子結晶解析結果 (2.57 Å) と一致する17)。しかし、H原子はGlu46に所属していた (図2)。フーリエ変換型赤外分光 (FTIR) による解析では、Glu46のprotonateに対応するC=O伸縮バンド (1740 cm–1近辺) が検出されており16)、今回のQM/MM計算結果と一致する。
(ポテンシャルエネルギー曲線) Freyによれば「H原子が2つのO原子から等距離にある場合は (LBHBではなくむしろ、さらに強いとされる) single-well H bond (結合長=2.4–2.5 Å) 」である19)。一方、OGlu46–OpCA結合距離2.57 Åは、single-well H bond の結合長とされる2.4–2.5 Åに対しては明らかに長すぎ5,6)、single-well H bondではあり得ない。つまり、「OGlu46–OpCA結合距離はLBHBだが、その水素原子位置はsingle-well H bond」という状況である。QM/MM計算によりOGlu46–OpCAのポテンシャル形状を調べたところ、single-well H bondでもLBHBでもない、通常の水素結合に見られるようなasymmetric double-wellポテンシャルの形状であった17) (図4)。
(NMR) OGlu46–OpCAのNMRのδH値は15.2 ppm20) と測定されており、LBHB (δH=17–19 ppm6)) としては低すぎる。実際、QM/MM構造からはδH=14.5 ppmと計算されたが、中性子構造からでは19.7 ppmであった21)。後者は、実験値15.2 ppm20) とかけ離れている。そもそも、「OGlu46から1.21 Å、OpCAから1.37 Å」(=single-well Hbondにみられる距離差0.16 Å) ならば、Limbachらの経験式22) からもマレイン酸並みに高い~20 ppmという値が予測される。 (逆に、ドナー・アクセプターが同じpKaを持つマレイン酸のような分子で初めてこのような位置にH原子を配置できる。)
(pKa) 過去の研究よりGlu46とpCAはそれぞれprotonate、deprotonateし、その結果としてpKa (Glu46) (=~9) >pKa (pCA) (=~6) であることが既知である13-16)。この傾向はQM/MM計算からも明らかではあるが (図4)、electrostatic titration計算からもPDB:2ZOHの構造ではpKa (Glu46) =8.6、pKa (pCA) =5.4であった17)。LBHBの条件「pKa値の一致」1-4) は満たされていないようである。

(Arg52のdeprotonation) 蛋白質内部ではsolvationenergyが得られないため、塩基性残基もdeprotonateし得る23,24)。しかし、蛋白質表面に露出しているArg52がdeprotonateしていること11) が事実だとしたら、(LBHBの存在以上に) これまでの蛋白質一般における研究結果の解釈の変更を求めるほどの大発見である。PDB:2ZOHの構造11) に対してQM/MM計算を行ったところ、Arg52をdeprotonateさせた場合、結晶構造中にあるArg52–Thr50間の水素結合 (2.93 Å) は崩壊して (3.78 Å) しまうのに対し、protonateの場合はほとんど変化がなかった (3.14 Å)。さらに、結晶構造座標とのRMSD (平均二乗偏差) においては、protonateの場合0.14 Åであるのに対し、deprotonateの場合0.35 Åとなり、結晶の分解能 (1.25 Å) から判断しても、deprotonated Arg52という解釈では無理が生じる17)。electrostatic titrationの結果でもArg52はprotonateしていた17)。そもそも、クロモフォアTyr42、Glu46、pCA上に存在する負電荷 (-1) が「孤立電荷で不安定」ならば、蛋白質は、静電相互作用の要請から自然とpCAをprotonationしてしまうことだろう。実際には、pCA上の負電荷は孤立電荷ではなく、Tyr42の-OHからの2.52 Å11) という非常に短い水素結合を受けていることは結晶構造からも明らかであり、不安定ではない (図2)。
5.おわりに
今回の中性子結晶構造、X線結晶構造は共に分解能が高く、データーの信頼性は極めて高いと思われる。一方で全ての水素原子が決定できたわけでもない。protonationサイトの水素原子が見られればprotonateしていると結論づけて良いが、逆は必ずしも真ではないため、より慎重な判断を要する。中性子結晶解析、NMR、FTIR、理論化学などをあわせて総合的な判断を下すことが、酵素反応機構解明およびその応用への近道になり得るだろう。
謝辞
本稿執筆に際し、恩師 長棟輝行先生 (東京大学) より機会を賜りました。心より感謝申し上げます。
文献
1) Perrin, C. L., Nielson, J. B.: Annu. Rev. Phys. Chem., 48, 511 (1997).
2) Cleland, W. W., Kreevoy, M. M.: Science, 264, 1887 (1994).
3) Frey, P. A., Whitt, S. A., Tobin, J. B.: Science, 264, 1927 (1994).
4) Schutz, C. N., Warshel, A.: Proteins, 55, 711 (2004).
5) Jeffrey, G. A.: An Introduction to Hydrogen Bonding, Oxford University Press, Oxford (1997).
6) Frey, P. A.: in Isotope Effects in Chemistry and Biology (Kohen, A. and Limbach, H.-H., Eds.) , 975, CRC press, Boca Raton, FL (2006).
7) Ishikita, H., Saenger, W., Biesiadka, J., Loll, B., Knapp, E.-W.: Proc. Natl. Acad. Sci. USA, 103, 9855 (2006).
8) Ishikita, H.: J. Biol. Chem., 282, 25240 (2007).
9) Anderson, S., Crosson, S., Moffat, K.: Acta Crystallogr. D Biol. Crystallogr., 60, 1008 (2004).
10) Fisher, S. Z., Anderson, S., Henning, R., Moffat, K., Langan, P., Thiyagarajan, P., Schultz, A.: J. Acta Crystallogr. D Biol. Crystallogr., 63, 1178 (2007).
11) Yamaguchi, S., Kamikubo, H., Kurihara, K., Kuroki, R., Niimura, N., Shimizu, N., Yamazaki, Y., Kataoka, M.: Proc. Natl. Acad. Sci. USA, 106, 440 (2009).
12) Borgstahl, G. E., Williams, D. R., Getzoff, E. D.: Biochemistry, 34, 6278 (1995).
13) Kim, M., Mathies, R. A., Hoff, W. D., Hellingwerf, K. J.: Biochemistry, 34, 12669 (1995).
14) Xie, A., Hoff, W. D., Kroon, A. R., Hellingwerf, K. J.: Biochemistry, 35, 14671 (1996).
15) Demchuk, E., Genick, U. K., Woo, T. T., Getzoff, E. D., Bashford, D.: Biochemistry, 39, 1100 (2000).
16) Kandori, H., Iwata, T., Hendriks, J., Maeda, A., Hellingwerf, K. J.: Biochemistry, 39, 7902 (2000).
17) Saito, K., Ishikita, H.: Proc. Natl. Acad. Sci. USA, 109, 167 (2012).
18) Ishikita, H.: J. Biol. Chem., 283, 30618 (2008).
19) Frey, P. A.: Magn. Reson. Chem., 39, S190 (2001).
20) Sigala, P. A., Tsuchida, M. A., Herschlag, D.: Proc. Natl. Acad. Sci. USA, 106, 9232 (2009).
21) Saito, K., Ishikita, H.: Biochemistry, 51, 1171 (2012).
22) Limbach, H.-H., Tolstoy, P. M., Pérez-Hernández, N., Guo, J., Shenderovich, I. G., Denisov, G. S.: Isr. J. Chem., 49, 199 (2009).
23) Ishikita, H., Knapp, E.-W.: J. Am. Chem. Soc., 129, 1210 (2007).
24) Ishikita, H.: FEBS Lett., 584, 3464 (2010).
![]()