�y�g�s�b�N�X�z
�����xEQCM�@�𗘗p�����^���p�N���ԓd�q�ړ��̑���ƍy�f������͂ւ̗��p
���q���s
�P�D�͂��߂�
EQCM (Electrochemical Quartz Crystal Microbalance) ����Ƃ́A�d�C���w����Ɛ����U���q�}�C�N���o�����X�@ (QCM�@) �Ƃ̓�������@�ł���1)�B�d�C���w����͓d�ɕ\�ʂł̕��q�̎_���Ҍ��̑���ł���B����AQCM�@�͓d�ɕ\�ʂ̏d�ʂ̌v���@�ł���A���q�̓d�ɕ\�ʂւ̋z������ђE���Ȃǂ��ʂ���B���������āA�������ɑ��肷�邱�Ƃɂ��A���q�̎_���Ҍ��������i�s����ۂ̋��������A���^�C���Ōv�����邱�Ƃ��\�ƂȂ�B����ɁAEQCM����������x�����邱�Ƃɂ��A�y�f�������̐��̔�������͂��邱�Ƃ��ł���B�y�f�Ɗ�Ƃ̔����ɂ����āA�d�ɏ�ɌŒ艻������̎_���Ҍ������Ɠ����ɍy�f-������̂̌`�����x�ׂ邱�Ƃ��\�ƂȂ�A����܂ŁA�n�t�̔������x�_�ł͑���ł��Ȃ��������J�j�Y���𖾂炩�ɂł���B
�d�q�ړ��ƊW�����y�f�����́A�~�g�R���h���A�̓d�q�`�B�n�A��������PSI�APSII���ł݂���B�~�g�R���h���A�̓����ł́A�^���p�N���ԓd�q�ړ������̘A�����i�s���Ă���A�d�q�ړ��̍ۂ̃|�e���V�����G�l���M�[�̃G�l���M�[ (ATP�ANADH�Ȃ�) �ɕϊ����Ă���B���ꂼ��̃^���p�N���̎_���Ҍ����S�͓S�����N���X�^�[��w���Ȃǂ̕⌇���q���ł���B
�����xEQCM����́A�����̓d�q�ړ��Ɛ��̔����̖��ڂȊ֘A���𖾂炩�ɂ���Ƌ��ɑf�����̃��J�j�Y������͂��邽�߂̗L���Ȏ�@�ł���B
�Q�D�^���p�N���̓d�C���w�ƍ����xEQCM����
�^���p�N���̓d�q�ړ��̌����́A�d�C���w����𗘗p�����������ʂ������A�L���Ȏ�@�̈�ł���B�������A�^���p�N���̓d�C���w����͍��̂Ƃ���e�Ղł���Ƃ͌����������B�^���p�N���̑���ɂ����ẮA��������������Ă��܂����� (���n�t���ł��邱�ƁA�����t�߂�pH�����A���x�����Ȃ�)�A�܂��A�^���p�N���̎_���Ҍ����S�ł���⌇���q���̓^���p�N�������ɑ��݂���̂ŁA�K�������d�ɂƓd�q�̎n�����ł��Ȃ��Ƃ�������肪����B����ɁA�d�ɕ\�ʂւ̋z���ɂ��^���p�N�����ϐ����邱�Ƃ������B����܂łɁA���d�ɕ\�ʂ̋@�\���A�Y�f�d�ɂ�p�����v���e�C���t�B�����@�ȂǁA���܂��܂ȍH�v�ɂ��^���p�N���̓d�C���w����@�͐i�����Ă���A�����̃^���p�N���̓d�q�ړ��ɂ��Ē��ׂ邱�Ƃ��o����悤�ɂȂ��Ă���2-6)�B
�}1��EQCM���葕�u�̊T���������B�d�C���w����̍�p�d�ɂ𐅏��U���q�̋��\�ʂƂ��A���莎���n�t�ƐڐG������B�����U���q�̐U�������v�����Ȃ���A��p�d�ɓd�ʂ�ω������邽�߂ɁA��p�d�ɂɂ̓|�e���V�I�X�^�b�g�Ǝ��g���J�E���^�[�̗������ڑ������B����ɂ��A�{���^���O�����ƐU�����ω����ɑ���ł���B�U�������d�ʊ��Z����ƁA��{�U����9 MHz�̐����U���q�ɂ����ĐU����1 Hz�̌�������1.4 ng�̏d�ʑ����ɑ�������B
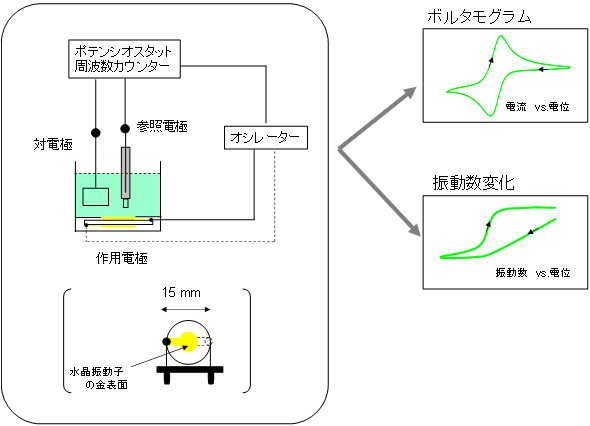
�}1�@EQCM���葕�u�̊T��
�������A���n�t���ł�EQCM����ł́A���x���Ⴍ�A10 ng���x�̕ω��𑪒肷��̂����E�ł���A100 ng���琔µg�̕ω����L�ӂȑ���ł���Ƃ���Ă����B���������āA�_���Ҍ��ɂ��d�������|���}�[�̏d�ʂ̑����A�_���Ҍ��ɂ������������̐͏o��z���̉�͓��ɗ��p����Ă���7,8)�B
����܂ŁA�^���p�N���̑���ւ̉��p���Ȃ���Ȃ������̂́A��ɏq�ׂ����x�̒Ⴓ�����ł������B�����ŁA�����x�����s���A�^���p�N���̑���ɉ��p�����B�����x���́A�d��Ǝ���̐U���m�C�Y�̏����ɂ���ĒB�����ꂽ�B�����U���q�̐U���ɂ�萶����U������ƃI�V���[�^�[�̋��U��H��������U������Ƃ̊����ŏ����ɂ��邽�߂ɁA���ꂼ��Ɨ��ɃV�[���h���A�m�C�Y�̒ጸ��}�����B���̌��ʁA���S�{�����x�����サ�A0.01 Hz���x���̕ω��𑪒肷�邱�Ƃ��\�ƂȂ����B����ɂ��A�^���p�N����EQCM���肪�\�ƂȂ�A����܂Ō��邱�Ƃ̂ł��Ȃ��������ۂ���͂ł���悤�ɂȂ����B
�R�D�����xEQCM����ɂ��V�g�N����c3�̓d�q�ړ��@�\�̉�
3-1�@�����i�K�ƂȂ�d�q���^�̃��J�j�Y��
�^���p�N���Ԃ̓d�q�ړ��̓����͌��܂������肩��d�q����e���A���܂�������ɓd�q���^���邱�Ƃł���B����������ƁA��������d�q�ړ����s���邱�Ƃł���B����Ɋւ��āA����܂łɒ��ږ��炩�ƂȂ��Ă��Ȃ��B�������A�������̎_���Ҍ��^���p�N���ł́A�d�q�����ƁA��莞�ԓd�q���^�ł��Ȃ��s���Ȏ��Ԃ����݂��邱�Ƃ������I�ɖ��炩�ɂ���Ă���B���̎��Ԃ����q�ԓd�q�ړ��̗����i�K�ƂȂ邱�Ƃ��������Ă���B���Ȃ킿�A�d�q����e����ƈ�莞�ԓd�q��ۗL���A�d�q���^���Ȃ��̂ł��낤�ƍl�����Ă���B
����܂łɁA[3Fe-4S]�N���X�^�[��L����t�F���h�L�V���ɂ����āA[3Fe-4S]�N���X�^�[�̃v���g�l�[�V�������d�q�ړ��ɑ��đ��x�𐧌����Ă��邱�Ƃ�����Ă���2)�B�d�ɂƃt�F���h�L�V���Ƃ̓d�q�ړ��ׂ����ʁA�t�F���h�L�V�����d�q��e�ɗv���鎞�Ԃ́A��1 ms�ł��邪�A�d�q���^����ɂ͖�30 ms�K�v�Ƃ��邱�Ƃ����炩�ɂ���Ă���B
���̂悤�ȓd�q��ێ����郁�J�j�Y���͖{���ɂ���̂ł��낤���B����ׂ邽�߂ɂ́A�^���p�N���̕��q�ԓd�q�ړ��ƕ��q�Ԃ̕����̌`���ڑ��肵�A���炩�ɂ���K�v������B
3-2�@�����xEQCM�ɂ��V�g�N����c3�̓d�q�v�[���@�\�̉�
�V�g�N����c3�͕��q���Ƀw��4��L����^���p�N���ł���B���_�Ҍ��ۗR���̃V�g�N����c3�̗��̍\�����}2�Ɏ����B���̓��ł́A�d�q�`�B�̂Ƃ��ē����A�����̓d�q���n������B�T�^�I�ȗ�ł́A���_�Ҍ��ۂɂ����āA�y�f�q�h���Q�i�[�[�����f�̎_��������G�}����Ƃ� (H2 �� 2H+ + 2e-)�A2�d�q���Ɏ��ƍl�����Ă���B
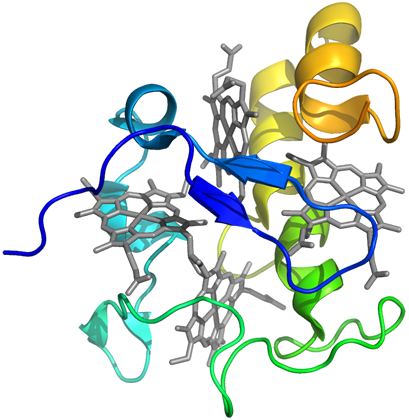
�}2�@�V�g�N����c3�̗��̍\��
�V�g�N����c3�̓d�q�ړ��̃��J�j�Y���Ƃ��āA�d�q�v�[���@�\�̑��݂������ꂽ�B�d�q�v�[���@�\�Ƃ́A�V�g�N����c3��4�d�q��e����ƁA����4�d�q��ێ������� �܁A��莞�ԓd�q���^���Ȃ��@�\�ł���9)�B��ɏq�ׂ��t�F���h�L�V���̏ꍇ�Ƃقړ��l�ł��邪�A�����̓d�q����������ɂ��A�d�q�v�[���ƌĂ��B
�d�q�v�[���@�\�́A�����xEQCM����ɂ�薾�炩�ɂ��ꂽ�B�}3�Ɏ����悤�ɁA�d�ɏ�ɌŒ艻���ꂽ�d�q�`�B�̂ƃV�g�N����c3�Ƃ̕��q�ԓd�q�ړ������n���\�z���A�_���Ҍ��Ɠ����ɐi�s���镡���̌`�������j�^�[����B����̊T�����}3�̖��ɂ��������Đ�������B
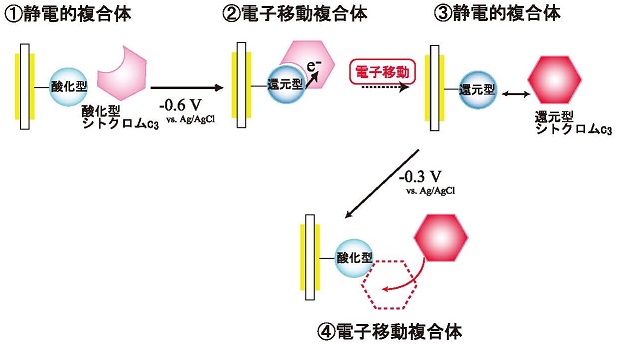
�}3�@�����xEQCM�𗘗p�������q�ԓd�q�ړ������̉��
�ŏ��ɁA�}3�̇@�ɂ���悤�Ȏ_���^�̌Œ艻���q�Ǝ_���^�̃V�g�N����c3�Ƃ̐Ód�I�ȕ����̂��`��������B�����ŁA�d�ɓd�ʂ�������ƁA�Œ艻���q���Ҍ��^�ƂȂ�A�_���^�V�g�N����c3�ւ̓d�q�ړ����n�܂�B���̂Ƃ��A�܂��A�d�q�ړ������̂��`������ (�A)�A���ɁA�d�q�ړ����i�s����B�d�q�ړ������̂ł�2���q���ڋ߂��邽�߁A�Ód�I�����̂̏ꍇ�����U��������������Ɨ\�z�����B�܂�A�V�g�N����c3���d�ɂɋ߂Â��̂ŁA�U��������������B�d�q�ړ����i�s����ƁA�B�ɂ���悤�ɁA�Ҍ��^�V�g�N����c3�ƌŒ艻���q�Ƃ̐Ód�I�����̂ɂȂ�B���ɁA�ĂьŒ艻���q���_���^�ɖ߂��A�Ҍ��^�V�g�N����c3����Œ艻���q�ւ̓d�q�ړ��������N�����B���̂Ƃ��A�Ҍ��^�V�g�N����c3���d�q��ێ�����Ȃ�A�d�q�ړ������̇C�̌`�����x���x���Ȃ�B�ȏ�̕��@�ŁA�d�q�ړ��ƕ����̌`���Ƃ̊W�ׂ邱�Ƃ��ł���B�Œ艻���q�ɂ́A���`���r�I���[�Q���U���̂𗘗p�����B���`���r�I���[�Q���U���̂́A�V�g�N����c3�̐��̓��ł̃p�[�g�i�[�ł͂Ȃ����V�g�N����c3�Ɠd�q�ړ��\�Ȏ_���Ҍ��d�ʂ�L���A���ۂɓd�q���ł���11)�B�d�q�v�[���@�\�́A�u�����������I������܂œd�q��ێ�����v���J�j�Y���ł��邩��A����𑪒肷��ɂ́A�V�g�N����c3�ɂƂ��āu�������Ȃ�����v��d�q�ړ��p�[�g�i�[�ɗp���Ȃ��Ă͂Ȃ�Ȃ��B���������āA�l�H�̎_���Ҍ������ł���r�I���[�Q���ނ��K���Ă���B
�}4�ɍ\�z�����d�ɂ̕\�ʂƃT�C�N���b�N�{���^���O�����̌��ʂ������B�}4 (a) �ɂ���悤�ɁA�d�ɏ�Ƀs���W���P���q�w���\�z���A���̏�ɒY�f��6�̃��`����������ăr�I���[�Q�����Œ艻�����B�r�I���[�Q���̌Œ艻���q����1.3 x10-11 mol cm-2�ł���B����̓T�C�N���b�N�{���^���O��������Z�o���� (�}4 (b) �j��)�B���ɂ��̃r�I���[�Q���\�ʂɃV�g�N����c3��Ód�I�Ɍ��������A�V�g�N����c3�̒P���q�w���\�z�����B�Ód�I�Ɍ��������V�g�N����c3�̕��q��7.4 x10-13 mol cm-2�͐����U���q�̐U��������Z�o�����B���̒l���猩�ς���ƁA�ׂ荇���V�g�N����c3�̋�����40 nm������A�V�g�N����c3���m���G�ꍇ�����͂قƂ�ǂȂ��B���������āA���̏����ł́A�r�I���[�Q���Ƃ̑��ݍ�p�݂̂��V�g�N����c3�̓����̋쓮�͂ɂȂ�B
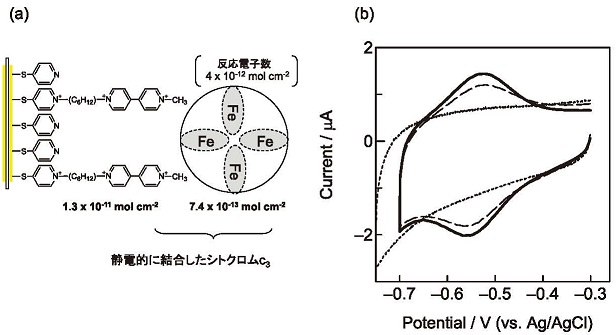

�}4 (b) �̃T�C�N���b�N�{���^���O��������V�g�N����c3�̎_���Ҍ��Ɋ֗^����d�q�����Z�o����ƁA4 x10-12 mol cm-2�ł���A�V�g�N����c3��4�`5�{�ɑ�������B���������āA�V�g�N����c3����4�̃w���S�Ă��_���Ҍ����Ă���B�܂��A�Z�o�����d�q���̓T�C�N���b�N�{���^���O�����̑|�����x�Ɉˑ����Ă��Ȃ����Ƃ���A�����̉��߂����������Ƃ��킩��B
���̓d�ɂƍ����xEQCM����ɂ��d�q�v�[���@�\�𑨂��邱�Ƃ��ł���B�d�ɓd�ʂ��X�e�b�v�����A���̂Ƃ��̐U�����ω����ɑ��肵�� (�}5)�B�d�ɓd�ʂ�-0.3 V����-0.6 V (vs.Ag/AgCl) �ɃX�e�b�v�����A�Œ艻���ꂽ�r�I���[�Q�����_���Ҍ�������B�Œ艻���ꂽ�r�I���[�Q����-0.3 V�ɂ����Ď_����ԁA-0.6 V�ł͊Ҍ���Ԃł���B
����0�b�ɂ����āA-0.3 V����-0.6 V�ւ̓d�ʃX�e�b�v���s���ƁA�Ҍ��^�r�I���[�Q������V�g�N����c3�ւ̓d�q�ړ��ߒ��A���Ȃ킿�A�V�g�N����c3�̓d�q��e�̗l�q��m�邱�Ƃ��ł���B���̂Ƃ��̐U�����ω�������ƁA�U�������f�����������A���悻0.5 s�ň��l�ɂȂ����B���̌��ʂ���A�d�q��e�̍ۂɂ́A�������ܓd�q�ړ������̂��`�����d�q�ړ����������Ă���Ƃ�����B���������āA�_���^�̃V�g�N����c3�͗e�Ղɓd�q����邱�Ƃ��ł���Ƃ�����B
���ɁA����5�b�ɂ����āA-0.6 V�������d�ɓd�ʂ�-0.3 V�ɕω��������B�����ł́A�Ҍ��^�V�g�N����c3����r�I���[�Q���ւ̓d�q�ړ��A�܂�A�V�g�N����c3�̓d�q���^�̃��J�j�Y����m�邱�Ƃ��ł���B�U�����́A�㏸������A�������ƌ��������l�ɂȂ�X�����������B���̌����ߒ����d�q�v�[���@�\�̑��݂m�ɕ\���Ă���B�d�ʂ�-0.6 V�ɂȂ����u�Ԃ̐U�����̏㏸����A�Ҍ��^�V�g�N����c3�Ǝ_���^�r�I���[�Q�����������Ă��邱�Ƃ�������B���Ȃ킿�A�d�q�ړ������̂͌`�����ꂸ�A�Ҍ��^�V�g�N����c3�͓d�q�����^���Ă��Ȃ� (�}5���̖͎��}�Q��)�B���̏�Ԃ��d�q�v�[����Ԃł���B
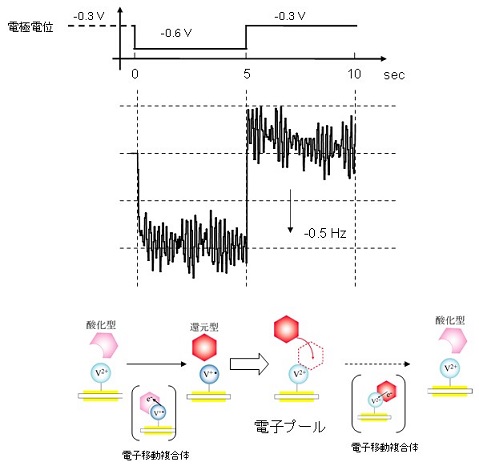
�}5�@EQCM����ɂ��d�q�v�[���@�\�̉��
�Œ艻���ꂽ�r�I���[�Q���̕��q���̓V�g�N����c3�̖�20�{�ł���A�܂��A�_���Ҍ��d�ʂ���l���Ă��A�e�Ղɓd�q�ړ��ł���B����ɂ��ւ�炸�A�����̂̌`�����ɒ[�ɒx�����Ƃ���A�Ҍ��^�V�g�N����c3�̓r�I���[�Q����F�����Ă��Ȃ����Ƃ��킩��B
�}5�̌����Ȑ�����Z�o�������x�萔��0.4 s-1�ł��邩��A�d�q�v�[����Ԃ̎�����2.5 s�ł���B��ʂɁA�^���p�N���Ԃ̓d�q�ړ������́A�x���Ƃ����x�萔�ɂ���100 s-1�̔����ł��邩��A�d�q�v�[����Ԃ̎����͑����F������ɏ\���Ȏ��Ԃł���B�d�q�v�[����Ԃ��o�R���邱�Ƃɂ���āA�����������F�����A�d�q�ړ����i�s���Ă���Ƃ�����B���ꂪ�A��������d�q�ړ����x���郁�J�j�Y���̈�ł���B
3-3�@�y�f�q�h���Q�i�[�[�Ɗ�V�g�N����c3�Ƃ̔���
�y�f�q�h���Q�i�[�[�́A�v���g���̊Ҍ�����ѐ��f�̎_����G�}����y�f�ł���B���_�Ҍ��ۓ��ł́A�d�q�`�B�^���p�N���V�g�N����c3�Ɠd�q������s���Ă���B�V�g�N����c3�͕��q���Ƀw����4�L����^���p�N���ł���B�v���g���̊Ҍ������ɂ����ẮA�Ҍ��^�̃V�g�N����c3����q�h���Q�i�[�[�֓d�q�ړ����i�s���A����A���f�̎_�������ł́A�q�h���Q�i�[�[����_���^�V�g�N����c3�ւ̓d�q�ړ����i�s����B�����̐G�}�����ɂ����āA���q�Ԃ̓d�q�`�B�̃��J�j�Y�����𖾂��邽�߂ɍs����EQCM�����������B
�܂��A�V�g�N����c3�Œ艻�d�ɂ����A�V�g�N����c3�̎_���Ҍ���d�ɂŃR���g���[������B�}6�ɌŒ艻�V�g�N����c3�̊T�O�}�������B���[�ɃA�W�h������w�L�T���`�I�[����p���ċ��\�ʏ�ɒP���q�w���\�z�����B�V�g�N����c3��N���[�ɂ̓A���L���𑤍��ɗL�����V�R�A�~�m�_�����Ă���A�d�ɏ�̃A�W�h�ƃN���b�N�����Ō���������10)�B���̌��ʁA�V�g�N����c3�̃w���P���d�ɑ��A�w��4���n�t���Ɉʒu��������ŌŒ艻���ꂽ���Ƃ��킩��B
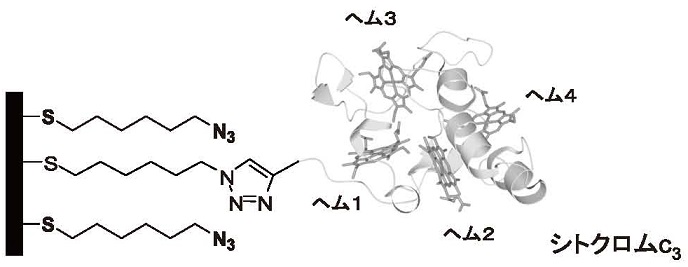
�}6�@�V�g�N����c3�Œ艻�d�ɂ̊T�O�}
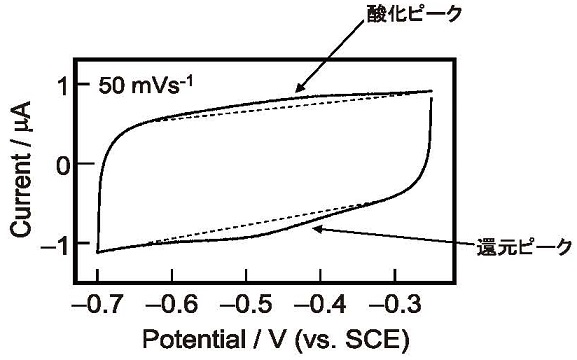
�}7�ɂ��̃V�g�N����c3�Œ艻�d�ɂ̃T�C�N���b�N�{���^���O�����������B�d�ɓd�ʂ͖O�a�J�������d�� (SCE) �ɑ��ĕ\�L�����B����ꂽ�{���^���O�����ɂ́A�_���s�[�N����ъҌ��s�[�N�������A�Œ艻�����V�g�N����c3���_���Ҍ����Ă��邱�Ƃ��킩��B�܂��A�{���^���O�����̎_���s�[�N����ъҌ��s�[�N����A�P�ʖʐϓ�����̌Œ艻���q�����Z�o�������ʁA�ׂ荇���V�g�N����c3���m�̋�����30 nm�ł��邱�Ƃ��킩�����B

�V�g�N����c3�ƍy�f�q�h���Q�i�[�[�Ƃ̔�����EQCM�ő��肷�邽�߂ɁA�V�g�N����c3�Œ艻�d�ɏ�ɍy�f�q�h���Q�i�[�[�̕��q�w���\�z�����B�V�g�N����c3�Œ艻�d�ɂƐڐG���Ă���ɏՉt�ɁA�q�h���Q�i�[�[�n�t��������ƁA�Ód�I�Ȍ����ɂ��A�V�g�N����c3��Ƀq�h���Q�i�[�[�̕��q�w���\�z�����B�Ód�I�Ɍ��������q�h���Q�i�[�[�̕��q������Z�o����ƁA�ق�1��1�̕����̂��`�������B
EQCM����ł́A���̐Ód�I�ȕ����̂��V�g�N����c3�̎_���Ҍ��ɂ���ĕω�����l�q�𑪒�ł���B�܂�A�Ód�I�����̓����ł̍y�f�q�h���Q�i�[�[�̓�����m�邱�Ƃ��ł���B
�}8��EQCM����̌��ʂł���B15�b�����ɓd�ɓd�ʂ�ω������A����Ɠ����ɋN����U�����̕ϓ����L�^�����B�d�ɓd�ʂ́A-0.6 V (vs.SCE) ����-0.3 V (vs.SCE) �̊Ԃŕω������A���ꂼ��A15�b�ԓd�ʂ�ۂ����B-0.3 V����-0.6 V�ɕω��������ꍇ�A�Œ艻���ꂽ�V�g�N����c3�͎_���^����Ҍ��^�ɂȂ�B���������āA�V�g�N����c3����q�h���Q�i�[�[�ւ̓d�q�ړ����i�s���� (���f���������̂̌`��)�B����A-0.6 V����-0.3 V�ɓd�ʂ�ω��������ꍇ�A�Œ艻���ꂽ�V�g�N����c3�͎_���^�ւƕω�����B���������āA�q�h���Q�i�[�[����V�g�N����c3�ւ̓d�q�ړ����i�s���� (���f�z�������̂̌`��)�B�}8�̊T�O�}�ɂ���悤�ɁA�V�g�N����c3�̎_���Ҍ���Ԃɂ��A�q�h���Q�i�[�[�ƌ�������ʒu���ς��ƍl������B���������āA�d�ʂ̃R���g���[���ɂ��A���f���������̌`���Ɛ��f�z�������̌`�������ꂼ��Ɨ��Ɋϑ����邱�Ƃ��ł���B����������ƁA���ꂼ��̕����̌`�������̋쓮�͂�Ɨ��ɗ^���Ă���ƕ\���ł���B���ꂪ�n�t���̔������x�_�I��͂ƈقȂ镔���ł���A�܂��A�G���g���s�[�ω��Ɉˑ������������������̂܂܊ϑ��ł���Ƃ���EQCM����̗��_�ł���Ƃ�����B
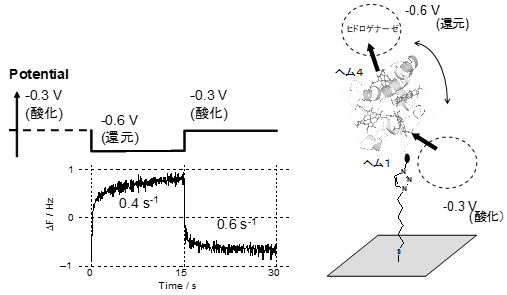
�}8�@EQCM���茋�ʂƔ������J�j�Y���̊T�O�}
�}8�ɂ����āA-0.3 V����-0.6 V�ɕω��������ꍇ������ƁA�U�������㏸���A���̌���l�ɂȂ��Ă��邱�Ƃ��킩��B����1 Hz�ȓ��̂����킸���ȐU�����̏㏸����A�q�h���Q�i�[�[���d�ɂ��牓�����������Ƃ��킩��B�q�h���Q�i�[�[�̓V�g�N����c3�ƐÓd�I�����̂��`�����Ă���̂ŁA���̐U�����ω��͕����̓����ł̃q�h���Q�i�[�[�̓����ł���B���������āA�Œ艻�����V�g�N����c3�ƃq�h���Q�i�[�[�͓d�ɂ��牓���ʒu�ŕ����̂��`�����Ă��邱�Ƃ��킩��B�܂��A�U�����ω��̋Ȑ����畡���̌`�����x�萔��0.4 s-1�ł������B����A-0.6 V����-0.3 V�ɕω��������ꍇ������ƁA�U�������������Ă��邱�Ƃ��킩��B����́A�q�h���Q�i�[�[���d�ɋߖT�ɋ߂Â��Ă������Ƃ������Ă���A�T�O�}�ɂ���悤�ɁA�����̌`���͓d�ɂɋ߂��ʒu�ōs���Ă���Ƃ킩��B
�Œ艻�V�g�N����c3�̔z������A�n�t���̓w��4�A�d�ɑ��̓w��1�ł���B���������āAEQCM����̌��ʂ���A���f���������̏ꍇ�́A�w��4�ߖT�ŕ����̂��`�����A���f�z���̏ꍇ�̓w��1�ߖT�ŕ����̂��`�����Ă��邱�Ƃ��킩��B�ȏ�̂悤�ɁA�����xEQCM����ɂ��A�d�q�ړ����y�f�����q���x���ʼn�͂��邱�Ƃ��ł���B
�S�D������
�y�f�̋@�\���𖾂����ŁA���p�I�ȓd�C���w����͗L�p�ȕ��@�ł���B�������Ȃ���A�ᕪ�q�̔�����͂ɗ��p�ł������@���y�f�����̑���ɗ��p�ł��Ȃ��ꍇ�������B����Љ���d�C���w��������ł���EQCM����́A�����x���ɐ������A�^���p�N���A�y�f�̑���ɗ��p�ł����B����A�l�X�ȃ^���p�N���A�y�f�̔�����͂��s�����Ƃɂ��A�y�f�̓d�q�ړ������̉𖾂��i�ނƍl������B
����
1) Buttry, D. A., Ward, D. M.: Chem. Rev., 92, 1355 (1992).
2) Chen, K., Hirst, J., Camba, R., Bonagura, C. A., Stout, C. D., Burgess, B. K., Armstrong, F. A.: Nature, 405, 814 (2000).
3) Murgida, D. M., Hidebrant, P.: J. Pys. Chem. B, 105, 1578 (2001).
4) Avila, A., Gregory, B. W., Niki, K., Cotton, T. M.: J. Phys. Chem. B, 104, 2759 (2000).
5) Khoshtariya, D. E., Wei, J., Liu, H., Yue, H., Waldeck, D. H.: J. Am. Chem. Soc., 125, 7704 (2003).
6) Gomes, I., Di Palolo, R. E., Pereira, P. M., Pereira, I. A. C., Saraiva, L. M., Penades, S., Franco, R.: Langmuir, 22, 9809 (2006).
7) Buttry, D. A., Ward, D. M.: Chem. Rev., 92, 1355 (1992).
8) Tasuma, T., Yokoyama, Y., Buttry, D. A., Oyama, N.: J. Phys. Chem. B, 101, 7556 (1997).
9) Asakrua, N., Kamachi, T., Okura, I.: J. Biol. Inorg. Chem., 9, 1007 (2004).
10) Iida, S., Asakura, N., Tabata, K., Okura, I., Kamachi, T.: Chem. Bio. Chem., 7, 1853 (2006).
11) Sanghoon, S., Asakura, N.: Electrochem. Commun., 34, 161 (2013).
![]() �@
�@