【トピックス】
大腸菌における新規エステル生産のための経路デザイン
田代洋平、渥美正太
理研・環境資源科学研究センター、カリフォルニア大・デービス・化学
1.はじめに
低炭素社会へ向けて、微生物を用いたバイオマスからの物質生産技術が、その注目度を高めている1)。微生物生産における最大のボトルネックのひとつは、出発物質から目的生産物までの代謝経路を有した微生物を得ることである。伝統的には、自然環境サンプルからの単離、あるいは育種によって獲得されてきたが、それらのプロセスは多くの時間と労力を要する。また、獲得した菌体の培養が困難である場合も少なくない。近年では、膨大に蓄積された代謝経路や酵素情報を基に、所望の代謝経路を人工的にデザイン、そして構築することが主流になりつつある。実際に、大腸菌や酵母といった扱いやすい宿主をベースとして、燃料やバルクケミカル,医薬品など有用化合物の生産菌が次々と創出されている2)。そのような分野は代謝工学と呼ばれ、本稿では、筆者らのエステル生産に関する取り組み3)を例に挙げて、その代謝工学アプローチについて紹介したい。
エステルとは、有機酸とアルコールから成る化合物であり、側鎖の長さによってその特性が異なる。低分子のカルボン酸エステルは揮発性や芳香性を有し、果実や花から発せられる香りに多く含まれている。それらのエステルは、動物や昆虫の誘引や病原菌からの防衛など植物にとって生理的な役割も果たしている。我々の生活においては、食品や香味料、化粧品の芳香、工業的には溶剤やペンキなどその用途は多岐にわたる4)。2012年、エステルの世界市場は166億ドルにも達した。近年では、ジェット燃料の代替燃料として利用可能なエステルも見いだされており5)、各用途に応じたエステルをバイオマスから高い生産性で合成できれば、二酸化炭素削減への貢献も見込まれる。
2.エステル合成経路のデザイン
2-1 熱力学的に有利なエステル合成反応
高効率なエステル合成経路を実現する上で、熱力学的に不利な反応は避けるべき対象である。筆者らは、まず、そのエステルの合成メカニズムに着目した。化学工業プロセスにおいて、エステルは有機酸とアルコールの脱水縮合によって合成される。常温・水相において、エステルの脱水縮合反応よりも、その逆反応である加水分解反応が熱力学的に有利であるため、既存のエステル合成プロセスでは、高温条件、さらに酸触媒を用いることによって、その平衡をエステル合成へとシフトさせる必要がある4)。エステラーゼやリパーゼなどの酵素を利用したとしてもこの熱力学的な不利は解消できない。
酵母などの細胞は、ビールやワインの発酵プロセスにおいて、短・中鎖エステルを生産することが知られている6)。これらの細胞では、有機酸の代わりにアシルCoAが有機酸ユニットとして用いられる (図1a)7)。この反応では、アシルCoAのアシル基が、アルコールO-アシル転移酵素 (ATF) によって、アルコールの水酸基へ転移されることでエステルが生成される。この反応の利点は、アシルCoAのチオエステル結合に蓄えられた高いエネルギーを利用できることにある。チオエステル結合の加水分解によって生じる自由エネルギーは、カルボン酸エステル結合のそれよりも大きいため (ΔG =-7.5 kcal/mol vs ΔG = -5 kcal/mol)、アシルCoAからアルコールへのアシル転移反応は、熱力学的に有利である。この反応を利用すれば、たとえ常温・水相条件下でも、高効率なエステル生産が可能となる。この原理は,酵素を用いたワックスエステルの生産にも利用されてきた。筆者らは、これを炭素数4から14までの短・中鎖エステルの生産に応用した3)。
2-2 エステル合成経路のコンビナトリアルデザイン
目的のエステルに応じて、その前駆体となるアルコールおよびアシルCoAの供給経路がそれぞれ必要とされる。既報において、様々な種類のアルコール合成経路が構築され、そのいくつかは高効率な経路となっている8)。一方、大腸菌におけるアシルCoAの供給経路は、物質生産を目標として工学された例はなかった。
細胞において、最も豊富なアシルCoAは炭素数2のアセチルCoAであり、アセチルCoAを有機酸ユニットとして用いた場合、酢酸エステルが生成される (図1b)。自然界において、分岐アミノ酸やリグニンの分解経路、あるいはイソプレノイドの代謝経路の中間体として分岐アシルCoAや芳香性アシルCoAが生成されることが知られており9,10)、これらの経路を大腸菌で利用できれば、より複雑なエステルも合成可能となる。理論的には、生産可能なエステルの種類は、アシルCoA供給経路とアルコール合成経路の組み合わせ数となる (図1b)。アシルCoA供給経路とアルコール合成経路のモジュール化しておけば、それらのモジュール経路を組み合わるだけで、所望のエステル合成経路を構築できる。
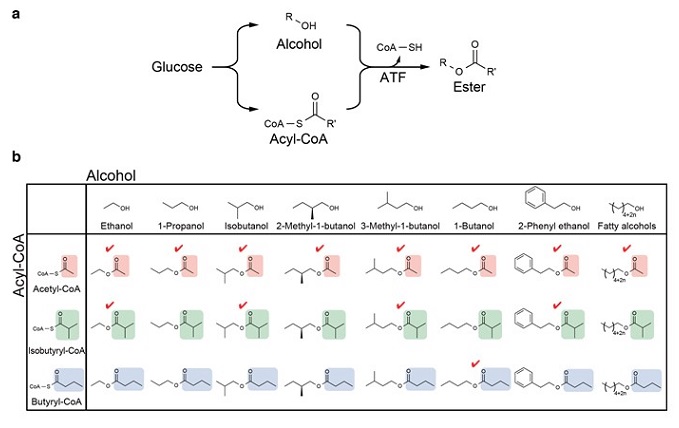

3.酢酸エステル合成経路の構築
3-1 α-ケト酸からの酢酸エステル合成
酢酸エステルは、酢酸とアルコールから成るエステルである。酢酸エステルの合成には、アセチルCoAが酢酸ユニットの代わりに必要とされるが、大腸菌においてアセチルCoAは豊富に存在する。そのため、大腸菌にアルコール合成経路とそれに適したATFを導入すれば、酢酸エステル合成経路を構築できる。
筆者らは、まずα-ケト酸ベースのアルコール合成経路に着目した。α-ケト酸は、α-ケト酸脱炭酸酵素およびアルコールデヒドロゲナーゼ (ADH) によって,高効率でアルコールへと変換することができる11)。筆者らは、α-ケト酸から酢酸エステルへの3ステップの経路を大腸菌にデザインした。α-ケト酸からアルデヒドまでの経路を強化するために、筆者らは2つの酵素遺伝子 (乳酸菌Lactococcus lactis由来のα-ケト酸脱炭酸酵素 (KDC) と出芽酵母Saccharomyces cerevisiae由来のATF (ATF1)) を大腸菌へ導入した。最終反応のアルデヒドデヒドロゲナーゼに関しては、大腸菌はもともと10種類以上のアルデヒドデヒドロゲナーゼを保有しており、多様なアルデヒドをアルコールへ高効率で変換することができるため、ここではadhの追加発現は行わなかった3)。この株の培養液に、種々のα-ケト酸を加え、37℃で振とうしたところ、加えたα-ケト酸から予想される酢酸エステルが生産された。この結果から、ATF1が分岐アルコールに対する寛容な基質選択性および高いアセチル化活性を有することが分かった。
つぎに、ATF1の直鎖アルコールに対する活性を評価するため、同株の培養液に炭素数2から10の直鎖アルコールを添加し、酢酸エステルの合成量を調べた。酢酸エチルや酢酸ブチルなどの短い酢酸エステルの合成量は高くなかったものの、炭素数6よりも長い鎖長の酢酸エステルは高濃度で検出された。これは、炭素数6よりも長い直鎖アルコールはATF1のよい基質となることを示している。β酸化経路や脂肪酸生合成経路をベースとして、炭素数6以上の直鎖アルコール合成経路が構築されており8)、これらのアルコール合成経路とこのATF1の組合せでも高効率で酢酸エステルを生産できることが示唆された。
3-2 グルコースからの酢酸イソブチルの生産
筆者らは、α-ケト酸-酢酸エステル経路を拡張し、グルコースからの酢酸エステルの合成経路の構築に取り組んだ。目的生産物として、酢酸イソブチルを選択した。酢酸イソブチルの前駆体となるイソブタノールは、グルコースから高効率で生産できることが示されており11)、このイソブタノール合成経路をベースとすれば、高収率・高収量の酢酸イソブチル合成が期待できる。イソブタノール合成能を向上させるために、5つの酵素遺伝子 (Bacillus subtilis由来のアセト乳酸合成酵素 (als)、大腸菌由来のケトール酸レダクトイソメラーゼ (ilvC) およびジヒドロキシ酸デヒドラターゼ (ilvD)、L. lactis由来のα-ケト酸脱炭酸酵素 (kdc) およびアルコール脱水素酵素 (adh))、さらにイソブタノールを酢酸イソブチルへ変換するためにATF1を大腸菌へ導入した。この株を用いて、グルコースから酢酸イソブチルの生産を試みた。
この大腸菌株は、24 h培養の時点で2.7 g/Lの酢酸イソブチルを生産したが、その後、その生産性は大きく低下した。最終的な細胞密度は、24 hの時点と比較して、20%減少していた。大腸菌の酢酸イソブチルに対する耐性濃度を調べたところ、およそ3 g/Lであったことから、生産性の低下は酢酸イソブチルの毒性によるものと推察された。
筆者らは、in situ 抽出により、酢酸イソブチルの収量および収率の向上を目指した。有機溶媒を用いる二層培養法は、中鎖アルコールやモノテルペノイド化合物など細胞毒性の高い化合物生産でとられる方法のひとつである12,13)。また、in situ 抽出によって、水相における最終生産物を低濃度に保つことは、その生産の駆動力にもなる14)。本研究では、抽出相として、細胞毒性の低くかつ疎水性の高いヘキサデカンを選択した13)。この二層培養法では、イソブタノールはその高い水溶性のため培養液相に存在するが、酢酸イソブチルへ変換されると疎水性が高くなりヘキサデカン相へと移動する (図2)。この二層培養法では、酢酸イソブチルの合成量は、24 h培養時点で、単層培養の時の合成量を超え、3.9 g/Lへ達した (図2)。その後も培養を続けると、72 hの時点で、酢酸イソブチル合成量は17.2 g/Lにも達した。このときの収率は理論最高収率のおよそ80%であった。
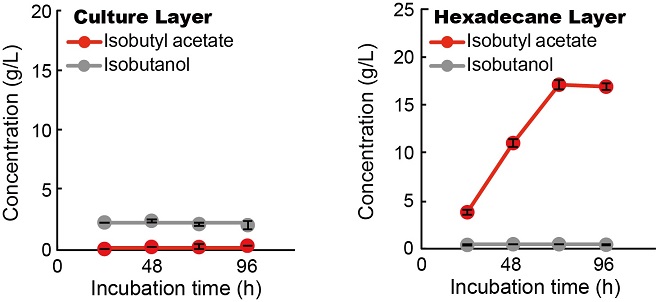
![]()
3-3 グルコースからの酢酸テトラデシルの生産
アルキル鎖の鎖長が14から18の酢酸エステルは化粧品の潤滑剤として利用され、その高いエネルギー密度のため燃料としても期待されている2)。これまで、脂肪酸の生合成経路やβ酸化経路をベースとして、直鎖アルコール合成経路が構築されてきた15-18)。本研究で提案するエステル合成戦略は、直鎖アルコールのエステル変換へも応用できる。
Vibrio harveyiというグラム陰性菌はある条件を満たすと発光を呈することで知られ、その発光メカニズムが明らかにされている19)。この発光機能を担うルシフェラーゼは直鎖の炭素数14のアルデヒド (テトラデカナール) を基質とする。そのテトラデカナールは、LuxCDE酵素複合体によって、脂肪酸生合成経路の中間体のひとつであるテトラデシリルACPから生成する (図3a)19)。筆者らは、LuxCDE酵素を大腸菌に発現させたところ、その株はテトラデカノールを生産することを見いだした (図3b)。大腸菌内在性の脂肪アルデヒドデヒドロゲナーゼによって、LuxCDEに生成されたテトラデカナールはテトラデカノールへと変換されたものと考えられる。筆者らは、大腸菌内でこのテトラデカノール経路に、ATF1を追加導入することにより、最大で140 mg/Lの酢酸テトラデシルの生物生産に成功した (図3bおよびc)。
以前より、脂肪酸エチルがディーゼルエステルとして注目され、その代謝工学が大腸菌などで行われてきた20,21)。脂肪酸エチルは、エタノールと炭素数16-18アシルCoAからエステルは作られる。酢酸テトラデシルを構成するアルコールおよび有機酸における鎖長の関係は、脂肪酸エチルのそれと逆転している。その構造的特徴から酢酸テトラデシルもよいバイオ燃料のターゲットと成りうる22)。
これらディーゼルエステルの合成では、アルコールおよびアシルCoAが1:1で生産されることが理想的である。しかし、アルコールとアシルCoAはどちらも、アセチルCoAを出発物質として生成されるため (図3a)、そのバランスの制御は困難である。筆者らが構築した酢酸テトラデシル合成経路においては、ATF1の発現に強力な発現プロモーターを利用するとその酢酸テトラデシル生産はほぼ検出できなくなった。Zhangらは、脂肪酸エチルの生産性向上のため、細胞内のアシルCoAの濃度に応じて、エタノールと脂肪酸生合成経路の活性をダイナミックに制御するシステムを開発した23)。彼らはこのシステムによって、大腸菌において1.5 g/Lの脂肪酸エチル合成に成功している。このような発現制御システムを取り入れることにより、酢酸テトラデシルの生産性の向上も可能であろう。
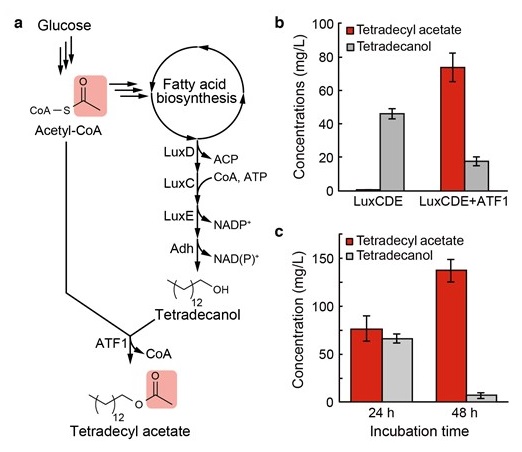

4.より高級なエステル合成経路の構築
4-1 大腸菌における分岐アシルCoA供給経路の構築
エステル合成経路を拡張するためには、より大きなアシル鎖をもつCoAの供給経路が必要となる。大腸菌において、β酸化経路の中間体として直鎖アシルCoAが生成されることは知られているが、分岐アシルCoAは生成されない。自然界において、ある細菌や菌類において、バリンやロイシンなど疎水的な分岐アミノ酸の分解経路の中間体として分岐アシルCoAが生成されることが報告されている9)。これらの分解経路では,α-ケト酸デヒドロゲナーゼ複合体 (KDHC) によりα-ケト酸から分岐アシルCoAが生成される。この反応メカニズムは、ピルビン酸デヒドロゲナーゼによるアセチルCoA生成と同様のものである。この反応では、α-ケト酸から不可逆的にアシルCoAが生成される。
筆者らは、Pseudomonas putida g7由来のKDHCオペロンを大腸菌に導入し、その活性を調べた。その結果、KDHCは、ピルビン酸よりも大きなα-ケト酸 (α-ケト酪酸、α-ケト吉草酸、α-ケトイソ吉草酸、α-ケト-3-メチル吉草酸、およびα-ケト-4-メチル吉草酸) に対して、高い活性を有することがわかった。これらの結果は、大腸菌にP. putida由来のKDHCを発現させることより、α-ケト酸から分岐アシルCoAの経路を構築できること示していた。
4-2 より高級なエステルの生産
P. putida KDHCを利用し、酢酸エステルよりも高級なエステルであるイソ酪酸エステルの合成を試みた。KDHCオペロンおよびATF1を大腸菌へ導入し、その株のイソ酪酸エステルの合成能を測定した。しかし、ATF1はアセチルCoAを基質として強く好むため、高級なエステルの合成には適していないことが明らかとなった (図4a)。そこで、出芽酵母のATF1以外のATF (EEB1およびEHT1) ,そしてクロラムフェニコールアセチル転移酵素 (Cat) の3つの酵素について、分岐アシルCoAに対する活性を比較した (図4a)。その結果、イソ酪酸イソブチルにはCat、イソ酪酸3-メチルブチル、イソ酪酸2-フェニルエチルおよび酪酸ブチルにはEHT1が、それぞれ適していることが分かった。イソ酪酸イソブチルに関しては、グルコースからの生産も可能であった3)。EHT1はエタノールヘキサノイルCoA転移酵素として知られているが、この結果から、EHT1はエタノールに対して高い選択性を有するわけではなく、より長鎖のアルコールも基質として認識することが示された。また、EHT1およびCatは、ジェット燃料の代替物質である酪酸ブチルの合成にも有用であった3)。
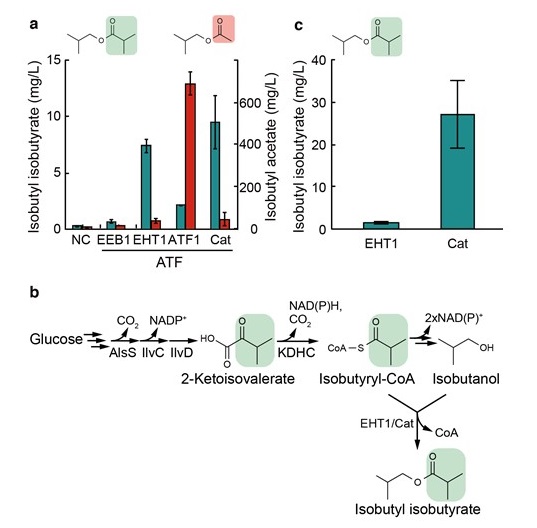

本研究の他にも高級エステルの生産のための研究が報告されている。Laytonらは、酪酸エステル合成のために、大腸菌に、β酸化回路をベースとするブチリルCoA供給経路を構築している24)。彼らは、数十mg/Lの酪酸エチル、酪酸イソプロピル、そして酪酸イソブチルの生産に成功した。また、Guoらは、脂肪酸生合成経路とβ酸化経路を巧みに組み合わせ、炭素数12から18までの直鎖アシルCoAの供給経路を大腸菌に構築した25)。これらのアシルCoAによって、炭素数2から5までの直鎖および分岐アルコールをアシル化することにより、16種類もの脂肪酸エステルの合成に成功した。
いずれのエステル合成系においても、経路や酵素の組み合わせによって、ある程度、生産物選択性や生産性を変えることができている。しかしながら、酢酸エステルと比較して、改善の余地を残しているといえる。本研究における高級エステルの生産においては、高級エステルの10倍量のアルコールが未反応で残っていたことから、アシル化反応がエステル生産の律速であると考えられる。さらなる生産効率の向上には、アシルCoA供給経路およびアシル転移酵素の基質選択性および活性の改良が必須だろう。
5.おわりに
本研究では、アシルCoAを有機酸ユニットとして用いる戦略により、大腸菌の低分子エステル生産能を飛躍的に向上させた。また、アルコール合成経路とアシルCoA合成経路の組み合わせにより、多彩なエステルの微生物生産が可能になった。ATFとアルコール合成経路を注意深く組み合わせることによって、生産されるエステルの選択性をある程度制御できた。
エステル合成経路を例にとってみてきたように、商業化レベルの収量および収率を達成するためには、経路全体のデザイン、各酵素の反応機構、そして各酵素の活性を注意深く選択し、組み合わせる必要がある。特に、不可逆反応の有無は、重要な要素のひとつである。効率の高いイソブタノール合成経路においては、ALSおよびKDCが高い活性を有し、さらに、脱炭酸をともなう不可逆反応であることが、高収率および高収量の大きな要因であると考えている。本研究で構築した酢酸イソブチル合成経路では、重要な中間体であるアセチルCoAは、細胞においてはピルビン酸から不可逆な脱炭酸反応により合成される。さらに、イソブタノールのアシル化において、アセチルCoAにチオエステル結合として保存されたエネルギーを駆動力として利用している。これらも酢酸イソブチルの高収量・収率の要因であろう。今後、このような代謝経路のデザイン原理は、さらに重要視されるだろう。
謝辞
本研究は、University of California-Davis startup fund、およびHellman Fellowshipの支援を受けて行ったものである。田代洋平は、日本学術振興会の支援を受けていた。
文献
1) Ragauskas, A. J., Williams, C. K., Davison, B. H., Britovsek, G., Cairney, J., Eckert, C. A., Frederick, W. J., Jr., Hallett, J. P., Leak, D. J., Liotta, C. L., Mielenz, J. R., Murphy, R., Templer, R. Tschaplinski, T.: Science, 311, 484 (2006).
2) Rabinovitch-Deere, C. A., Oliver, J. W., Rodriguez, G. M. Atsumi, S.: Chem Rev, 113, 4611 (2013).
3) Rodriguez, G. M., Tashiro, Y., Atsumi, S.: Nat Chem Biol, 10, 259 (2014).
4) Iwasaki, T., Maegawa, Y., Ohshima, T., Mashima, K.: Kirk-Othmer Encyclopedia of Chemical Technology, 1 (2012).
5) Chuck, C. J., Donnelly, J.: Appl Energy, 118, 83 (2014).
6) Lilly, M., Bauer, F. F., Lambrechts, M. G., Swiegers, J. H., Cozzolino, D., Pretorius, I. S.: Yeast, 23, 641 (2006).
7) Fujii, T., Nagasawa, N., Iwamatsu, A., Bogaki, T., Tamai, Y., Hamachi, M.: Appl Environ Microbiol, 60, 2786 (1994).
8) Lan, E. I., Liao, J. C.: Bioresour Technol, 135, 339 (2013).
9) Mooney, B. P., Miernyk, J. A., Randall, D. D.: Annu Rev Plant Biol, 53, 357 (2002).
10) Bugg, T. D., Ahmad, M., Hardiman, E. M., Rahmanpour, R.: Nat Prod Rep, 28, 1883 (2011).
11) Atsumi, S., Hanai, T., Liao, J. C.: Nature, 451, 86 (2008).
12) Connor, M. R., Cann, A. F., Liao, J. C.: Appl Microbiol Biotechnol, 86, 1155 (2010).
13) Brennan, T. C., Turner, C. D., Kromer, J. O., Nielsen, L. K.: Biotechnol Bioeng, 109, 2513 (2012).
14) Shen, C. R., Lan, E. I., Dekishima, Y., Baez, A., Cho, K. M., Liao, J. C.: Appl. Environ. Microbiol., 77, 2905 (2011).
15) Howard, T. P., Middelhaufe, S., Moore, K., Edner, C., Kolak, D. M., Taylor, G. N., Parker, D. A., Lee, R., Smirnoff, N., Aves, S. J., Love, J.: Proc Natl Acad Sci USA, 110, 7636 (2013).
16) Lin, F., Das, D., Lin, X. N., Marsh, E. N.: FEBS J, 280, 4773 (2013).
17) Schirmer, A., Rude, M. A., Li, X., Popova, E., del Cardayre, S. B.: Science, 329, 559 (2010).
18) Dellomonaco, C., Clomburg, J. M., Miller, E. N. Gonzalez, R.: Nature, 476, 355 (2011).
19) Meighen, E. A.: FASEB J, 7, 1016 (1993).
20) Kalscheuer, R., Stolting, T., Steinbuchel, A.: Microbiology, 152, 2529 (2006).
21) Steen, E. J., Kang, Y., Bokinsky, G., Hu, Z., Schirmer, A., McClure, A., Del Cardayre, S. B., Keasling, J. D.: Nature, 463, 559 (2010).
22) Barney, B. M.: Nat Chem Biol, 10, 246 (2014).
23) Zhang, F., Carothers, J. M., Keasling, J. D.: Nat Biotechnol, 30, 354 (2012).
24) Layton, D. S., Trinh, C. T.: Metab Eng, 26C, 77 (2014).
25) Guo, D., Zhu, J., Deng, Z., Liu, T.: Metab Eng, 22, 69 (2014).
![]()