�y�g�s�b�N�X�z
�����Ǔ��i������������cDNA display�V�X�e��
��ʑ�E���H��
�P�D�͂��߂�
���q�i���𗘗p�����y�v�`�h��^���p�N�����q�f�U�C���̌����́A1985�N��G. P. Smith��ɂ��Phage display�@�̊J���Ɏn�܂�BPhage display�@�͌��݂ł��ł��L�͂ȃE�C���X�^��`�q�^�]�\���^�Ή��t���̐i���H�w��@�ł���A�R�̍H�w�ɂ��s���ȋZ�p�ƂȂ��Ă���B����A�{�e�ŏq�ׂ閳�זE�|��n�𗘗p�����E�C���X�^��`�q�^�]�\���^�Ή��t����display�Z�p�́A���C�u�����T�C�Y�̑傫�� (phage display��103�ȏ�) �ɉ�����V�R�A�~�m�_�����ɂ��Ή����邱�Ƃ���V�R�̌n�Ƃ͎����̈قȂ�L�͂Ȕz���Ԃ̒T�����\�Ƃ���B
1-1�@mRNA display�@��cDNA display�@
�����Ǔ��Ńy�v�`�h��^���p�N���̕��q�i���ɕK�{�Ȗ��זE�|��n�̎��p��������1988�N�Ƀ��V�A��Spirin��ɂ���ĕ��ꂽ1)�B���̕�[���Ƃ��A���זE�|��n��p������`�q�^ �] �\���^�Ή��t�����q�̃R���Z�v�g�������̐i���H�w�����O���[�v�����Ă��ꂽ�B�����āAribosome �����mRNA�Ƃ���ɃR�[�h���ꂽ�^���p�N����A������polysome display2)�Aribosome display3)���J������A�����ăs���[���}�C�V���𗘗p����ribosome�̉�݂Ȃ��Œ���mRNA�ƐL�����̃|���y�v�`�h�����L�����Œ��ژA��������mRNA display (�܂���in vitro virus)4,5)���J�����ꂽ�B������t�����̖��זEdisplay�Z�p�ł́A��������mRNA1���q�ɑ���1���q�̃|���y�v�`�h��A�����邽�߁A���́A���זE�|��n�̍��������͂���قǏd�v�łȂ����{�\�[���̃^�[���I�[�o�[�����A�ނ���mRNA�̕������傫�ȉۑ�ł������B�����mRNA display�ł�mRNA-�^���p�N���̌`�������͓���1�������ł���A�`���̑O��ƂȂ�mRNA��puromycin-linker��T4 RNA ligase�ɂ��A�������ł���18���Ԕ��������Ă�50���ȉ��ł������B�����ăA�N�����A�~�h�Q���d�C�j���ł̘A���Y���̐�o���������K�v�Ȃ��߁A���̎�@�͎��p�I�Ƃ͌���������B�t�@�[�W�f�B�X�v���C�Ɠ����̎��p����������ɂ́AmRNA�����̐v���ȋt�]�ʂɂ��cDNA�� (���艻) �ƌ����I�Ȓ����@�̊m�����K�v�ł���B����܂����������ł́Apuromycin-linker�ɗl�X�ȍH�v���{���ƂƂ��ɒ����ߒ���������邱�Ƃ�cDNA display (��q) ���J�������B���̕��@�ł�mRNA�̋t�]�ʂƓ����Ƀ|���y�v�`�h��cDNA�����L���������邽�� (�}1)�A�����̂̈��萫�̔���I����ƒ����菇�̊ȑf�������������B�܂��A�ʏ��in vitro selection�ł̓y�v�`�h�A�v�^�}�[�̌�₪�����Ă��A�@�\�̊m�F�̂��߂̌����A�b�Z�C�ɑ���Ȏ�ԂƎ��Ԃ��₷�̂��ʗ�ł���B�M�҂��cDNA display�ppuromycin-linker��p�����v���_�E���E�A�b�Z�C��\�ʃv���Y�������� (SPR) ��͂ɂ��A�v�����ȕւȋ@�\�]�����@���J������6,7)�B���Ƃ��X�N���[�j���O�v���Z�X�����������Ă��A�����������╪�q�̋@�\�A�b�Z�C�������ƂȂ邽�߃X�N���[�j���O���Ԃւ̊�^�͌���I�ł���B�܂�A��X��in vitro selection�����łȂ���╪�q�̌����I�ȃA�b�Z�C�@�����ǁE�����������邱�ƂŃX�N���[�j���O�V�X�e���Ƃ��Ă�cDNA dispay�Z�p�̊����x���I�ɍ��߂Ă����B
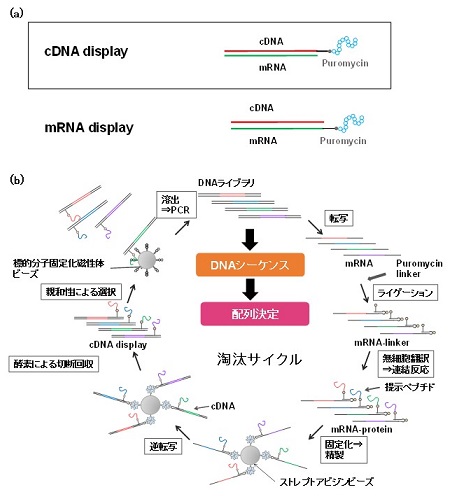

�Q�DcDNA display�V�X�e���̉ۑ�Ƃ��̍���
cDNA display�@�ɂ͓����A�����̃X�e�b�v�̑��i���ɔ���cDNA display���q�̎����̒Ⴓ�A�܂�����ꂽ���^���p�N���ɑ���n�C�X���[�v�b�g�]�� (���ݍ�p���) �@�̖��m���Ȃǂ̂��܂��܂ȉۑ肪�����͂������Ă����B�����ł͂��̎�ȉۑ�Ƃ����ɑ��ču������������Љ��B
2-1�@mRNA��Puromycin-linker�̘A������
cDNA display���q�́A�@mRNA��puromycin-linker�̘A���̂��疳�זE�|��n��p���Ă�mRNA display���q�̍쐻�A�A�����q��streptavidin (SA) �����r�[�Y�ł̐����Apuromycin-linker�̋t�]�ʃv���C�}�[�̈悩��mRNA�����̋t�]�ʂ��o�Ē��������BmRNA��puromycin-linker�̘A���́AmRNA��3’���[��linker��5’���[�Ƃ̍y�f�I�A���ɂ��BmRNA display�@���J�����ꂽ�����́Apuromycin-linker��ߏ艻�ł�T4 DNA���K�[�[�Ȃ���T4 RNA���K�[�[�ɂ��A�����s���Ă��� (�}2a)�B����ɑ�cDNA display�@�ł�mRNA��puromycin-linker���n�C�u���_�C�[�[�V���������邱�Ƃ�T4 RNA���K�[�[�̔������������߂�8)�B�n�C�u���_�C�[�[�V�����ɂ��A�������͑傫�����P�������̂́A������cDNA display�@�Ŏg��ꂽpuromycin-linker (long biotin-segment puromycin-linker�G LBP) �ł́AmRNA��LBP�̃�����1:4�ɂ�����1���Ԓ��x�̔�����v���� (�}2b)�B ����ɁA�y�f������ɐ������K�v�Ȃ��߃n�C�X���[�v�b�g�͖]�߂Ȃ��B������puromycin-linker�̍\�������ǂ��邱�Ƃɂ��short biotin-segment puromycin-linker (SBP) ���J���� (�}2c)9)�AmRNA��linker�̃�����1:1�ɂ�����10���̔�����90%�ȏ�̔������������������B�������Apuromycin-linker���c�����Ȃ����ߔ�����̐������s�v�ł���AmRNA��puromycin-linker�̘A�������t�ɖ��זE�|��n�ɓY�����邱�Ƃ��ł���悤�ɂȂ����B
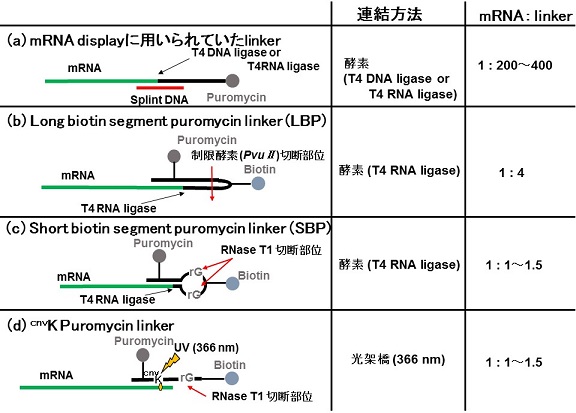

mRNA display�@�ł́AmRNA��puromycin-linker�̘A���ւ̉ˋ��� (�\������) �̊��p����������Ă���10)�Bpuromycin-linker��5’���[�Ƀ\�����������AmRNA��linker�̃n�C�u���_�C�[�[�V������Ɏ��O����30�����x�Ǝ˂����mRNA��linker�̘A���������炳���B���̕��@�͍y�f�Ɉˑ�����������ȒP�Ȃ��߁AmRNA�̔C�ӂ̉ӏ��ł̘A�����\�ł���ƂƂ��Ƀn�C�X���[�v�b�g�ł�����B�������\��������p�������@�ł́A���O�� (300 nm>) �Ǝ˂ɂ��mRNA�̑������s���ŁA�t�]�ʌ����̒ቺ������Ă���11)�B���̖����������ׂ����������ł͍ŋ߁A�k����[�Ȋw�Z�p��w�@��w�̓��{�炪�����������ˋ���3-Cyanovinylcarbazole (cnvK) ��p����puromycin-linker (cnvK linker) ���J������ (�}2d)12)�BcnvK linker�ł�1�����x�̎��O���Ǝ˂�mRNA��linker��f�����A���ł��邽�߁AmRNA�����̔���I�y����mRNA�����̗}�������������B�O�q�̂悤��cnvK��linker�̖��[�����łȂ��z����ɂ������ł��邱�Ƃ���Alinker�v�̎��R�x�����߂�ƂƂ��ɁA�y�f�i���Ȃǂ��܂��ܗp�r�ł�puromycin-linker�J���ւ̓W�]���J�����B
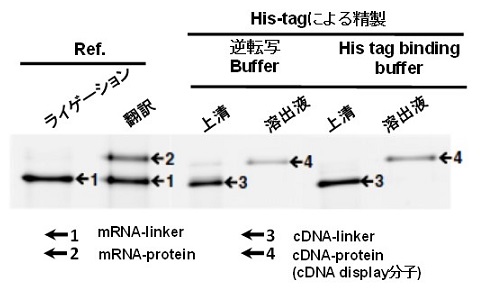
2-2�@cDNA display���q�쐻�̌������P
cDNA display���q�̒������̂̊ȕ��E�v�����͌��サ�Ă��邪�A���f���^���p�N���Ƃ���B-domain of protein A (BDA) ��p�����ۂ�cDNA display���q�̎����͓�������mRNA��puromycin-linker�̘A���̂̂킸��1�����x�������B��������̂��߁A�܂�mRNA display���q��SA�r�[�Y�ւ̌Œ艻���������������B���זE�|��ɂ���ē���ꂽmRNA display���q��mRNA�ɂ̓��{�\�[�������������܂܂ł���A���̃��{�\�[���ɋN�����闧�̏�Q��SA�r�[�Y�Ƃ̐ڋ߂�j��ł���Ɛ��@���ꂽ�B�����ŁA���זE�|���̗n�t��EDTA��������mRNA���烊�{�\�[�����𗣂��������mRNA display���q��SA�r�[�Y�ւ̌Œ艻�����݂��Ƃ���A�Œ�����͖�20�����サ���B�����90���ȏ�̌Œ������ɂ͕����l��100�{���x��SA�r�[�Y���K�v�ł��邱�Ƃ���������13)�B�����̍H�v�ɂ��A��90%�ȏ�̌�����mRNA display���q������ł���悤�ɂȂ����BcDNA display�@�ł́A�|���ƂꂽcDNA/mRNA-linker���q����菜���ړI��cDNA display���q�̃^���p�N��������His tag�����Ă���ANi-NTA�r�[�Y��p����cDNA display���q������B�]����SA�r�[�Y�����RNase T1�ɂ��E���ɂ�RNase T1�p��Buffer��p���Ă���A����Buffer��His tag�����Ɏ�������ł��܂��Ă����B�������ANi-NTA�r�[�Y�p��Buffer�ł�RNase T1�������������ꂽ���Ƃ���]�v��Buffer�̎�������������A�����̌���Ǝ菇�̊ȑf�����ł����B�����̉��P�ɂ��ABDA�ł�cDNA display���q�̍쐻�ɂ����������17%�Ɍ��サ�� (�}3)�B���Ȃ킿, �]����cDNA display�@�ɔ�ׂă��C�u�����T�C�Y�������I�Ɉꌅ�ȏ㑝�債�����Ƃ���A���p�I�ȕ��@�̈�ɒB�����B

2-3�@���זE�|��n��p�������q�ԑ��ݍ�p���
cDNA display�@�Ȃǂ̎����Ǔ����������ł͓�����ɕ����̌��z��DNA��������B�]���͓���ꂽ���z��DNA�̏�����ɑ咰�ۂł̔����≻�w�I�y�v�`�h�������o�Ă��̋@�\����͂��Ă������A���������ɔ�₷���ԂƃR�X�g���c��Ȃ����A�\�ʃv���Y���������͖@ (SPR�@) �Ȃǂɂ�鑊�ݍ�p��͂ɂ͉ߏ�ȗʂ������唼�͖��ʂɂȂ炴��Ȃ������B���̂��߁A���z��DNA����v���������ɓK�ʂ̃^���p�N����y�v�`�h���������A�@�\��͂ɋ������@���]�܂ꂽ�B�����œ��������ł�puromycin-linker�Ɩ��זE�|��n��g�ݍ��킹�����L�̕��@���J�����Ă����B
��߂͓������ꂽ�z��R���̃y�v�`�h��^���p�N���̕��q�ԑ��ݍ�p��萫�I�ɉ�͂���Pull-down�@�ł���B�܂��A����ꂽmRNA�ɐ�p��puromycin-linker��A�����Ė��זE�|��n��p���Ė|�邱�Ƃɂ����mRNA-linker-�^���p�N�������̂���������B���̌�AmRNA�����̕������o��linker����^���p�N���������̃r�[�Y�ɌŒ肷��B�����āA���̃^���p�N���Œ艻�r�[�Y��Pull-down�ɗp����B���Ȃ킿�A�W�I���q�̃^���p�N���Œ艻�r�[�Y�ւ̌����ʂ𑊑ΓI�ɕ]������ (�}4)6)�B�Ȃ��A�^���p�N���͎����r�[�Y�ɌŒ艻����Ă��邽�߁A�W�X���t�B�h�����≻�w�ˋ���L����^���p�N���ɑ��鑊�ݍ�p��͂��e�Ղł���14)�B���̎�@��p����A����ꂽ�z��̂����ł�������╪�q�Ƒ��ݍ�p���̂�Z���Ԃōi�荞�ނ��Ƃ��ł���B
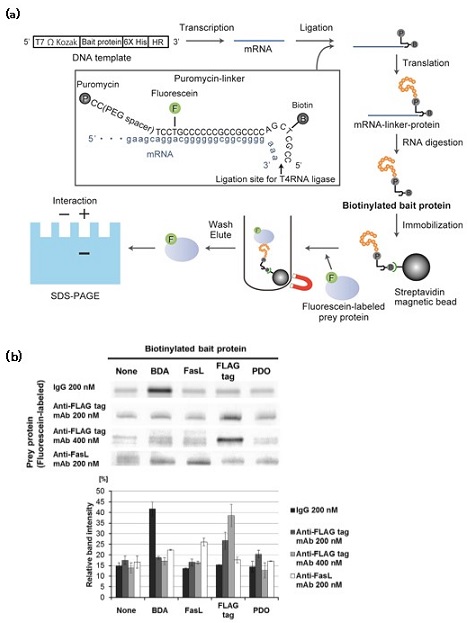

������́A�\�ʃv���Y�������� (Surface Plasmon Resonance; SPR) ���葕�u (Biacore) �ŕ��q�ԑ��ݍ�p���ʉ������@�ł���B���̎�@�ł́Apoly A�ƃr�I�`����z����puromycin-linker��p���A�|��Y�����I���SdT�r�[�Y�ɂ��������B���̌�A�Y����streptavidin�Z���T�[�`�b�v��ɌŒ艻���ASPR����p���K���h���q�Ƃ��đ��ݍ�p����͂���B���f���Ƃ��Ă�BDA��IgG�̑��ݍ�p����͂�mRNA�𖢕����̂܂܉�͂����Ƃ���A�����l����1���傫���𗣒萔�ƂȂ������AmRNA�����邱�Ƃŕ����l�Ɠ����̉𗣒萔������ꂽ (�}5)7)�B
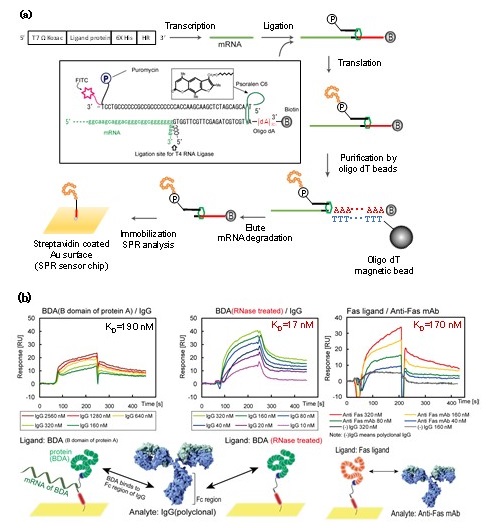

�咰�ۂł̃^���p�N�����≻�w�I�y�v�`�h�����Ƃ������]�����@�ł͌��ʂ�̂�1�`2������v���Ă����̂ɑ��A��q��2�̎�@�ł͂������1�`2���ʼn�͉\�ł���A�����Ǔ����������œ���ꂽ���z��̕��q�ԑ��ݍ�p�̐v����͂ւ̓������B
�R�D������
�s���[���}�C�V����DNA�����mRNA��A�������Ė��זE�|��n�Ŗ|�����mRNA�ƃ^���p�N�����A������Ƃ���in vitro virus (mRNA display) �@�̔������獡�N��20�N�ƂȂ�B���̊ԁA�����O�ɂ����đn��𒆐S�Ƃ��āA���̋Z�p�������x���`���[��Ƃ̐ݗ����������A���łɑ傫�Ȑ��ʂ���������B�������Ȃ���A�咰�ۂ�p�����t�@�[�W�f�B�X�v���C�@�Ɣ�����̈�`�q�H�w�̉����Ƃ��Ă͓���ʂ�����AmRNA display�@�͖�����ʓI�ȋZ�p�ɂ͎����Ă��Ȃ��B�ŋ߁AcDNA display�@�ւ̉��ǂ��Ȃ���Ĉ��萫�����܂�ƂƂ��ɁAcnvK�����J�[�̓����ɂ��v�����ƊȈՉ��������炳�ꂽ���ƂŁA���q�����w�n�̌������ł���C�y�Ɉ������Ƃ��ł���悤�ɂȂ����BcDNA display�V�X�e���̓t�@�[�W�f�B�X�v���C�̏��Ȃ��Ƃ�1000�{�̃��C�u�����T�C�Y��L����ƂƂ��ɁA��`�q�^ (cDNA) �ƕ\���^ (�^���p�N��) ���ɂ߂ĒP���ɋ��L�����������q�`��ł��邽�߁A�l�X�ȏ������ł̑I���ɂ��Ή��\�ł���B���������āA�n����u�������e�a���T�������łȂ��A�y�f�Ɋւ���G�}�@�\�T���ɂ����Ă��d�v�ȃc�[���ɂȂ肦��B����ɁA�L�@�n�}���Ŋ���������“�l�I�o�C�I���q”�Ƃł������ׂ����̍����q�̒T����̕����E��ʗL�@�������������ɂ�钴�������q�̑n���ȂǁA�A�C�f�A����Ŏ�X�̐V�K������J��ł�����̂Ɗ��҂����B
�ӎ�
cDNA display�Z�p�̉��ǂɑ���ȍv�������Ă����������k����[�Ȋw�Z�p��w�@��w�̓��{���������A�Y�ƋZ�p�������������ʌ������̖]���C�����m�A���̑��A���ʂ̓s���ŋL�ڂł��܂������̌������̊w���̕��X�ɐ[�����ӂ������܂��B�܂��A���̓x�̋M�d�Ȏ��M�̋@������������܂������{����w�̓����Y�����A�~�J���y�����ɂ��S��芴�Ӑ\���グ�܂��B
����
1) Spirin, A. S., Baranov, V. I., Ryabova, L. A., Ovodov, S. Y., Alakhov, Y. B.: Science., 242, 1162 (1988).
2) Mattheakis, L. C., Bhatt, R. R., Dower, W. J.: Proc. Natl. Acad. Sci. USA., 91, 9022 (1994).
3) Hanes, J., Plückthun, A.: Proc Natl Acad Sci USA., 94, 4937 (1997).
4) Nemoto, N., Miyamoto-Sato, E., Husimi, Y., Yanagawa, H.: FEBS Lett., 414, 405 (1997).
5) Roberts, R. W., Szostak, J. W.: Proc. Natl. Acad. Sci. USA., 94, 12297 (1997).
6) Mochizuki, Y., Kohno, F., Nishigaki, K., Nemoto, N.: Anal Biochem., 434, 93 (2013).
7) Nemoto, N., Fukushima, T., Kumachi, S., Suzuki, M., Nishigaki, K., Kubo, T.: Anal Chem., 86, 8535 (2014).
8) Yamaguchi, J., Naimuddin, M., Biyani, M., Sasaki, T., Machida, M., Kubo, T., Funatsu, T., Husimi, Y., Nemoto, N.: Nucleic Acids Res., 37, e108 (2009).
9) Mochizuki, Y., Biyani, M., Tsuji-Ueno, S., Suzuki, M., Nishigaki, K., Husimi, Y., Nemoto, N.: ACS Comb. Sci., 13, 478 (2011).
10) Kurz, M., Gu, K., Lohse, P. A.: Nucleic Acids Res., 28, E83 (2000).
11) Mayar, G.: Nucleic Acid and Peptide Aptamers Methods and Protocols, 535, 293 (2009).
12) Mochizuki, Y., Suzuki, T., Fujimoto, K., Nemoto, N.: J. Biotechnol., 212, 174 (2015).
13) Mochizuki, Y., Kumachi, S., Nishigaki, K., Nemoto, N.: Biol Proced Online. 15, doi: 10.1186/1480-9222-15-7. (2013).
14) Tanemura, Y., Mochizuki, Y., Kumachi, S., Nemoto, N.: Biology (Basel)., 4, 161 (2015).
![]() �@
�@