亂僩僺僢僋僗亃
Nonribosomal peptide synthetase 偵偍偗傞撪晹傾僨僯儗乕僔儑儞僪儊僀儞傪梡偄偨僕儁僾僠僪崌惉
埨晹抭巕丄彫椦鐫旻
搶嫗揹婡戝丒棟岺丄拀攇戝堾丒惗柦娐嫬
侾丏偼偠傔偵
婡擻惈僆儕僑儁僾僠僪偺桳塿惈偼惢栻嬈奅傗寬峃怘昳丄壔徬昳嬈奅側偳懡條側暘栰偱尋媶偝傟偰偍傝1-3)丄傑偨丄僆儕僑儁僾僠僪傪棙梡偡傞偙偲偼廬棃偺傾儈僲巁棙梡傪奼戝偝偣傞壜擻惈傗怴偨側傾儈僲巁梡搑傪惗傓壜擻惈傪傕偮丅備偊偵怴偨側峺慺朄偵傛傞僆儕僑儁僾僠僪崌惉朄偑拲栚偝傟偰偄傞4,5)丅杮僩僺僢僋僗偱偼丄変乆偑採埬偟偨丄nonribosomal peptide synthetases (NRPS) 偺1偮偱暋悢偺僪儊僀儞偐傜側傞儅儖僠僪儊僀儞峺慺撪偺 “internal (撪晹偺) ” 傾僨僯儗乕僔儑儞僪儊僀儞偺傒傪梡偄偨僆儕僑儁僾僠僪崌惉朄傪徯夘偡傞丅
俀丏傾僨僯儗乕僔儑儞僪儊僀儞偺摥偒
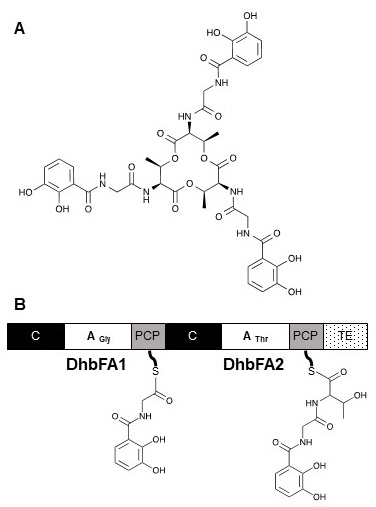
僶僔儕僶僋僠儞 (2,3-dihydroxybenzoate-glycine-l-threonine trimeric ester) (恾1A) 偼揝偑朢偟偄娐嫬壓偱揝僀僆儞傪廤傔傞偨傔偵僶僋僥儕傾偵傛偭偰崌惉偝傟傞僔僨儘僼僅傾偲屇偽傟傞壔崌暔偱偁傞6,7)丅僶僔儕僶僋僠儞偼nonribosomal peptide synthetases (NRPSs) 8) 偵傛偭偰崌惉偝傟傞丅NRPS偼僶僋僥儕傾偺擇師戙幱嶻暔偺丄庡偵彫偝側儁僾僠僪偺崌惉偵娭梌偟偰偍傝丄偙傟傜偺儁僾僠僪偺偄偔偮偐偼丄峈惗暔幙傗峈僂僀儖僗栻丄柶塽梷惂嵻丄嵶朎暘楐慾奞嵻側偳偲偟偰偺棙梡偑婜懸偝傟偰偄傞9,10)丅堦斒揑側NRPS偼丄婎幙偲側傞傾儈僲巁傪慖戰偟偰妶惈壔偟丄傾儈僲巁摨巑傪寢崌偟偰儁僾僠僪傪慻傒棫偰傞儔僀儞偲偟偰婡擻偡傞僞儞僷僋幙儐僯僢僩偺偄偔偮偐偺僙僢僩偐傜側傞丅偼偠傔偵傾僨僯儗乕僔儑儞僪儊僀儞偑aminoacyl-O-AMP傪崌惉偟丄偦偺屻丄peptidyl carrier protein (PCP) 僪儊僀儞偑傕偮4’-phosphopantetheine枛抂偺僠僆乕儖婎傊傾儈僲巁傪嫟桳寢崌偝偣傞丅Condensation僪儊僀儞偼儁僾僠僪嵔怢挿偺夁掱偱儁僾僠僪寢崌宍惉傪怗攠偡傞丅僶僔儕僶僋僠儞偺惗崌惉偵偍偄偰DhbF (恾1B) 偲偄偆儅儖僠僪儊僀儞峺慺偵娷傑傟傞2偮偺傾僨僯儗乕僔儑儞僪儊僀儞偼丄偦傟偧傟僌儕僔儞傑偨偼 L-僗儗僆僯儞傪妶惈壔偟丄PCP僪儊僀儞偵寢崌偟偨4’-phosphopantetheine偵寢崌偝偣傞栶妱傪傕偮11)丅杮尋媶偱偼丄偙偺DhbF偑傕偮2偮偺撪晹傾僨僯儗乕僔儑儞僪儊僀儞傪DhbFA1偁傞偄偼DhbFA2偲柦柤偟丄幚尡偵梡偄偨丅傑偨丄NRPS偺傾僨僯儗乕僔儑儞僪儊僀儞偼superfamily of adenylate-forming enzyme偲屇偽傟傞僗乕僷乕僼傽儈儕乕偵懏偟偰偍傝丄偙偺僗乕僷乕僼傽儈儕乕偵偼丄傾僔儖CoA崌惉峺慺傗傾僙僠儖CoA崌惉峺慺丄儕僌僯儞偺宍惉偵娭梌偡傞4-僋儅儖巁:CoA儕僈乕僛丄偝傜偵偼儂僞儖儖僔僼僃儔乕僛摍偑懏偟偰偄傞12-14)丅

俁丏撪晹傾僨僯儗乕僔儑儞僪儊僀儞扨懱偱偺敪尰偍傛傃惛惢
DhbF偺撪晹偺傾僨僯儗乕僔儑儞僪儊僀儞偩偗傪敪尰偝偣偰挷傋偨曬崘偼側偐偭偨偨傔丄偙傟傜偑婡擻偡傞偨傔偵昁梫側堚揱巕椞堟偼晄柧偱偁偭偨丅偦偺偨傔丄adenylate-forming enzyme偺僗乕僷乕僼傽儈儕乕偵懏偡傞懠偺峺慺傗丄撈棫宆偺傾僨僯儗乕僔儑儞僪儊僀儞偺傾儈僲巁攝楍偲嫟偵 DhbF偺傾儈僲巁攝楍傪梡偄偰僔乕働儞僗傾儔僀儊儞僩傪峴偄丄偦傟傪傕偲偵DhbF偺撪晹傾僨僯儗乕僔儑儞僪儊僀儞偩偗傪敪尰偝偣傞偨傔偺椞堟傪寛掕偟偨丅偦偺椞堟偺DNA傪PCR偱憹暆偟偨屻偵丄僐乕儖僪僔儑僢僋敪尰儀僋僞乕偵慻傒崬傒丄戝挵嬠傪梡偄偰偺戝検敪尰偍傛傃堚揱巕嶻暔偺惛惢傪専摙偟偨寢壥丄DhbFA1偁傞偄偼DhbFA2偺僪儊僀儞偺傒偺僞儞僷僋幙傪摼傞偙偲偵弶傔偰惉岟偟偨 (恾2)丅


係丏撪晹傾僨僯儗乕僔儑儞僪儊僀儞傪梡偄偨僕儁僾僠僪崌惉
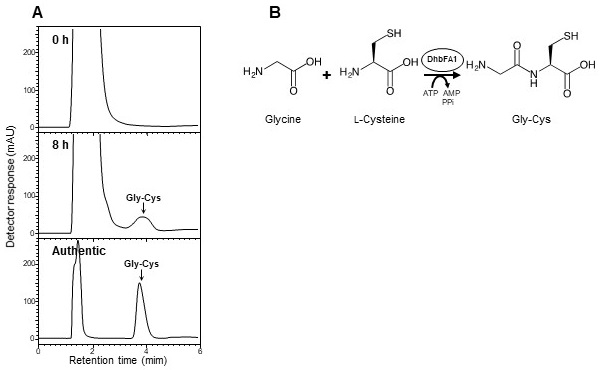
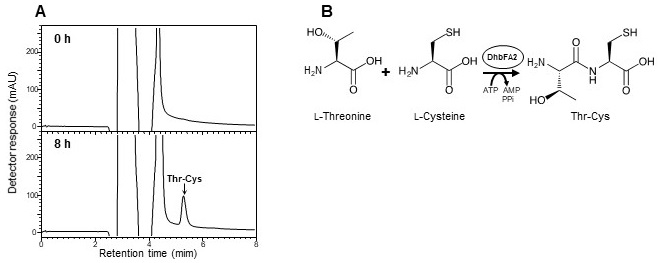
僶僔儕僶僋僠儞偺惗崌惉偵偍偄偰偼丄DhbFA1偼ATP傪梡偄偰僌儕僔儞傪妶惈壔偟丄PCP僪儊僀儞偵庴偗搉偡栶妱傪傕偮僪儊僀儞偱偁傞丅扨撈惛惢偟偨DhbFA1偵僌儕僔儞偲丄PCP僪儊僀儞偺戙傢傝偵 L-僔僗僥僀儞傪婎幙偲偟偰壛偊偰斀墳偝偣偨偲偙傠丄斀墳偺恑峴傪帵偡AMP検偺憹壛偑妋擣偝傟丄傑偨丄AMP埲奜偺嶻暔偺僺乕僋傕HPLC暘愅 (CROWNPAK CR (+) column, 4.0×150 mm, 堏摦憡梟攠 夁墫慺巁pH1.5) 偵傛傝妋擣偝傟偨 (恾3A)丅偙偺嶻暔偼丄暘庢丒惛惢屻丄幙検暘愅偵傛偭偰丄暘巕検偑178偱偁傞偙偲偑傢偐偭偨丅偙偺暘巕検偼僌儕僔儞偲L-僔僗僥僀儞偑扙悈偟偰寢崌偟偨暘巕検偲堦抳偡傞偑丄暘巕検偐傜梊憐偝傟傞斀墳嶻暔偲偟偰3偮偺壔崌暔 (僌儕僔儞偺僇儖儃僉僔儖婎偲L-僔僗僥僀儞偺僠僆乕儖婎偑僠僆僄僗僥儖寢崌偟偨壔崌暔S-glycyl-l-cysteine丄僌儕僔儞偺僇儖儃僉僔儖婎偲L-僔僗僥僀儞偺傾儈僲婎偑儁僾僠僪寢崌偟偨僕儁僾僠僪Gly-Cys丄偁傞偄偼偦偺媡偺L-僔僗僥僀儞偺僇儖儃僉僔儖婎偲僌儕僔儞偺傾儈僲婎偑儁僾僠僪寢崌偟偨僕儁僾僠僪Cys-Gly) 偑峫偊傜傟偨偨傔丄LC-MS/MS偵傛傞嶻暔偺摨掕傪峴偭偨丅偦偺寢壥丄僌儕僔儞偺僇儖儃僉僔儖婎偲L-僔僗僥僀儞偺傾儈僲婎偑儁僾僠僪寢崌偟偨Gly-Cys偵偟偐妋擣偝傟側偄僼儔僌儊儞僥乕僔儑儞 (m/z 99) 偑専弌偝傟丄撪晹傾僨僯儗乕僔儑儞僪儊僀儞偱偁傞DhbFA1偑僌儕僔儞偲L-僔僗僥僀儞傪寢崌偟偰僕儁僾僠僪傪崌惉偡傞偙偲傪弶傔偰柧傜偐偵偟偨 (恾3B)丅摨條偵丄扨撈惛惢偟偨DhbFA2偵L-僗儗僆僯儞偲L-僔僗僥僀儞傪婎幙偲偟偰壛偊偰斀墳偝偣偨応崌偵傕丄L-僗儗僆僯儞偲L-僔僗僥僀儞偑扙悈弅崌偟偨壔崌暔偲摨偠暘巕検傪傕偮嶻暔偺僺乕僋偑HPLC暘愅 (Develosil RPAQUEOUS column, 4.6×250 mm, 堏摦憡梟攠5%傾僙僩僯僩儕儖娷桳0.1%僊巁) 偱妋擣偝傟偨 (恾4A)丅LC-MS/MS 暘愅偺寢壥丄L-僗儗僆僯儞偺僇儖儃僉僔儖婎偲L-僔僗僥僀儞偺傾儈僲婎偑儁僾僠僪寢崌偟偨Thr-Cys偵偟偐妋擣偝傟側偄僼儔僌儊儞僥乕僔儑儞 (m/z 143) 偑専弌偝傟丄DhbFA2傕DhbFA1摨條偵僕儁僾僠僪傪崌惉偡傞偙偲傪柧傜偐偵偟偨 (恾4B)丅傑偨DhbFA1偱偼丄僌儕僔儞偺戙傢傝偵L-僙儕儞丄偁傞偄偼L-傾僗僷儔僊儞巁偲L-僔僗僥僀儞傪婎幙偲偟偰梡偄偨応崌偵傕妶惈偑妋擣偝傟丄Ser-Cys偍傛傃 Asp-Cys偺惗惉偑帵嵈偝傟偨丅


僕儁僾僠僪崌惉偵偍偗傞DhbFA1偁傞偄偼DhbFA2偺L-僔僗僥僀儞偵懳偡傞Km抣傪夝愅偟偨偲偙傠丄DhbFA1偺L-僔僗僥僀儞偵懳偡傞Km 抣偼22mM偲崅偐偭偨偑丄DhbFA2偺L-僔僗僥僀儞偵懳偡傞Km 抣偼0.016mM偲掅偄偙偲偑傢偐偭偨丅婎幙傾儈僲巁偵懳偡傞恊榓惈偑崅偄偙偲偼丄岺嬈揑側儁僾僠僪惗嶻偵偍偄偰廳梫偱偁傞丅
俆丏僆儕僑儁僾僠僪崌惉偲偦偺婡峔
DhbFA2偼Thr-Cys偺1庬椶偺僕儁僾僠僪偟偐崌惉偱偒側偐偭偨偑丄DhbFA1偼Gly-Cys埲奜偵傕L-僙儕儞傗L-傾僗僷儔僊儞巁傪娷傓Ser-Cys傗Asp-Cys傪崌惉偱偒傞偙偲偑帵偝傟偨丅偝傜偵丄DhbFA1傕DhbFA2傕丄L-僔僗僥僀儞偺戙傢傝偺婎幙偲偟偰N枛抂偵L-僔僗僥僀儞傪傕偮僆儕僑儁僾僠僪 (Cys-Gly傗Cys-Gly-Gly-Arg-Glu丄Cys-Gly-Gly-Arg-Glu-Ser-Gly-Ser-Gly-Ser) 傪梡偄偨応崌偵僩儕儁僾僠僪傗僿僉僒儁僾僠僪丄僂儞僨僇儁僾僠僪側偳偺僆儕僑儁僾僠僪傪崌惉偱偒傞偙偲偑帵偝傟偨丅偙傟傜偺僆儕僑儁僾僠僪偺崌惉妶惈偼丄L-僔僗僥僀儞傪婎幙偲偟偨僕儁僾僠僪偺崌惉妶惈偲偁傑傝曄傢傜側偐偭偨丅傑偨丄D-僔僗僥僀儞傕 L-僔僗僥僀儞偲摨條偵婎幙偵側傝偆傞偙偲偑傢偐偭偨丅偙傟傜偺偙偲偐傜丄N枛抂偑僔僗僥僀儞偱偁傟偽丄偳偺傛偆側傾儈僲巁攝楍偺儁僾僠僪偱傕婎幙偵偡傞偙偲偑偱偒傞偲峫偊傜傟傞丅
嬤擭丄傾僨僯儗乕僔儑儞僪儊僀儞偲摨偠偔adenylate-forming enzyme偺僗乕僷乕僼傽儈儕乕偵懏偡傞D-alanine:D-alanyl carrier protein ligase偱偁傞DltA偲偄偆峺慺傕儁僾僠僪傪崌惉偱偒傞偙偲傪変乆偼柧傜偐偵偟丄偦偺儊僇僯僘儉傪採彞偡傞傑偱偵帄偭偨16-18)丅DhbFA1偲DhbFA2傕adenylate-forming enzyme偺僗乕僷乕僼傽儈儕乕偵懏偡傞峺慺偱偁傞偨傔丄DltA偺儁僾僠僪崌惉婡峔傪傕偲偵変乆偑採埬偡傞DhbFA1偁傞偄偼DhbFA2偑娭梌偡傞怴婯儁僾僠僪崌惉斀墳偺婡峔偼師偺傛偆側傕偺偱偁傞丅 (i) 杮棃偺傾僨僯儗乕僔儑儞僪儊僀儞偺妶惈 (adenylation) 偵傛傝S-aminoacyl-cysteine偑拞娫懱偲偟偰宍惉偝傟丄懕偄偰 (ii) 壔妛揑側S→N傾僔儖揮堏斀墳偵傛傝N-aminoacyl-cysteine (儁僾僠僪) 偑崌惉偝傟傞 (恾5)丅偮傑傝丄峺慺揑側斀墳偲壔妛揑側斀墳偺擇抜奒偺楢懕偟偨斀墳傪宱傞儁僾僠僪寢崌偺宍惉偱偁傞丅DhbFA1偲DhbFA2偑忋婰偺儊僇僯僘儉偱儁僾僠僪傪崌惉偡傞偙偲偼丄DhbFA1偲DhbFA2偺偳偪傜偺峺慺偱傕丄僔僗僥僀儞偺戙傢傝偵傾儈僲婎傪帩偨偢偵僠僆乕儖婎偺傒傪傕偮僔僗僥僀儞傾僫儘僌偱偁傞 3-儊儖僇僾僩僾儘僺僆儞巁傪婎幙偲偟偰梡偄偨応崌偵偼斀墳偑恑傒丄僠僆乕儖婎傪帩偨側偄L-傾儔僯儞傗L-僙儕儞側偳偺傾儈僲巁傪婎幙偵梡偄偨応崌丄偁傞偄偼S-methyl-cysteine側偳偺曐岇偝傟偨僠僆乕儖婎傪傕偮壔崌暔傪婎幙偵梡偄偨応崌偵偼斀墳偑恑傑側偐偭偨偙偲偐傜傕峫嶡偝傟傞丅斀墳偵偼梀棧偺僠僆乕儖婎偑昁梫偱偁傝丄僇儖儃僉僔儖婎偑捈愙僔僗僥僀儞偺傾儈僲婎偲寢崌偡傞栿偱偼側偄偙偲傪帵偡丅偙偺斀墳儊僇僯僘儉偼Native chemical ligation (NCL) 朄19,20) 偲屇偽傟傞儁僾僠僪偺壔妛崌惉朄偺斀墳婡峔 (壜媡揑側僠僆乕儖-僠僆僄僗僥儖岎姺斀墳偺屻偵懕偄偰S→N傾僔儖揮堏斀墳偑婲偙傞) 偲椶帡偟偨揰偑偁傞偑丄変乆偑採埬偡傞曽朄偺戞1抜奒偺斀墳偼峺慺斀墳偱偁傞偨傔丄NCL朄偱偼昁梫側婎幙偺僠僆僄僗僥儖壔偑晄梫偲偄偆棙揰偑偁傞丅偟偐偟側偑傜丄戞2抜奒栚偺斀墳偼NCL朄偲摨條偱偁傞偨傔丄NCL朄偺墳梡斀墳傪嶲峫偵僆儕僑儁僾僠僪摨巑傪楢寢偡傞斀墳傗丄楢寢晹傪僔僗僥僀儞埲奜偺傾儈僲巁偵曄姺偡傞曽朄21)側偳傪尰嵼丄専摙拞偱偁傞丅
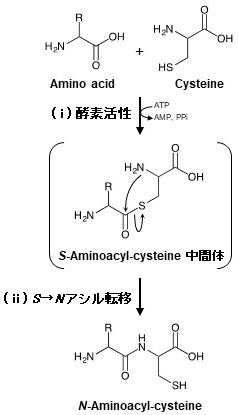
恾5丂傾僨僯儗乕僔儑儞僪儊僀儞偵傛傞怴婯儁僾僠僪崌惉斀墳婡峔
俇丏偍傢傝偵
杮尋媶偱変乆偼NRPS偺儅儖僠僪儊僀儞峺慺撪偺撪晹傾僨僯儗乕僔儑儞僪儊僀儞傪梡偄偰傕儁僾僠僪傪崌惉偱偒傞偙偲傪徹柧偟22)丄偙傟偼adenylate-forming enzymes偺僗乕僷乕僼傽儈儕乕偵娷傑傟傞峺慺慡偰偑晛曊揑偵傕偮妶惈偱偁傞偙偲傪嫮偔帵嵈偡傞傕偺偱偁傞丅杮僗乕僷乕僼傽儈儕乕偵偼條乆側婎幙摿堎惈傪傕偭偨峺慺偑娷傑傟傞偨傔丄杮僗乕僷乕僼傽儈儕乕偵懏偡傞峺慺傪梡偄傞偙偲偱桳梡側儁僾僠僪傪梕堈偵崌惉偡傞偙偲偑偱偒傞偩傠偆丅
幱帿
杮尋媶偼丄搶嫗揹婡戝妛棟岺妛晹惗柦棟岺妛宯 娐嫬旝惗暔妛尋媶幒傪拞怱偵峴傢傟偨尋媶偺惉壥偱偁傝丄尋媶幒偺奆條偵怱偐傜姶幱偄偨偟傑偡丅傑偨丄杮尋媶傪峴偆偵偁偨傝丄Bacillus subtilis strain168偺愼怓懱DNA傪暘梌捀偒傑偟偨拀攇戝妛 拞懞尠愭惗丄尋媶偵娭偟偰偛彆尵傪捀偒傑偟偨拀攇戝妛 嫶杮媊婸愭惗丄搶嫗揹婡戝妛 嶳柤徆抝愭惗偵姶幱怽偟忋偘傑偡丅杮尋媶偼暥晹壢妛徣偐傜偺壢妛尋媶曗彆嬥偺彆惉偺堦晹傪庴偗偨傕偺偱偡丅傑偨丄搶嫗揹婡戝妛憤崌尋媶強尋媶 (Q17L-02) 偲偟偰峴偭偨傕偺偱偡丅
暥專
1) Mills, S., Stanton, C., Hill, C., Ross, R. P.: Annu. Rev. Food Sci. Technol., 2, 299 (2011).
2) Santos, S., Torcato, I., Castanho, M. A.: Biopolymers, 98, 288 (2012).
3) Grottelli, S., Ferrari, I., Pietrini, G., Peirce, M. J., Minelli, A., Bellezza, I.: Int. J. Mol. Sci., 17, 1332 (2016).
4) Tanaka, T., Takagi, K., Saddam, H. M., Takeda, Y., Wakayama, M.: Appl. Biochem. Biotechnol., 183, 362 (2017).
5) Kino, H., Kino, K.: Biosci Biotechnol Biochem., 79, 1827 (2015).
6) Pi, H., Helmann, J. D.: Proc. Natl. Acad. Sci. USA, 114, 12785 (2017).
7) Abergel, R. J., Zawadzka, A. M., Hoette, T. M., Raymond, K. N.: J. Am. Chem. Soc., 131, 12682 (2009).
8) Finking, R., Marahiel, M. A.: Annu. Rev. Microbiol., 58, 453 (2004).
9) Labby, K. J., Watsula, S. G., Garneau-Tsodikova, S.: Nat. Prod. Rep., 32, 641 (2015).
10) Nguyen, K. T., Ritz, D., Gu, J. Q., Alexander, D., Chu, M., Miao, V., Brian, P., Baltz, R. H.: Proc. Natl. Acad. Sci. USA, 103, 17462 (2006).
11) May, J. J., Wendrich, T. M., Marahiel, M. A.: J. Biol. Chem., 276, 7209 (2001).
12) Gulick, A. M.: ACS Chem. Biol., 4, 811 (2009).
13) Ehmann, D. E., Shaw-Reid, C. A., Losey, H. C., Walsh, C. T.: Proc. Natl. Acad. Sci. USA, 97, 2509 (2000).
14) Conti, E., Franks, N. P., Brick, P.: Structure, 4, 287 (1996).
15) Röttig, M., Medema, M. H., Blin, K., Weber, T., Rausch, C., Kohlbacher, O.: Nucleic Acids Res., 39, W362 (2011).
16) Abe, T., Hashimoto, Y., Zhuang, Y., Ge, Y., Kumano, T., Kobayashi, M.: J. Biol. Chem., 291, 1735 (2016).
17) 埨晹抭巕, 嫶杮媊婸, 彫椦払旻: 僶僀僆僒僀僄儞僗偲僀儞僟僗僩儕乕, 74, 318 (2016).
18) Abe, T., Hashimoto, Y., Sugimoto, S., Kobayashi, K., Kumano, T., Kobayashi, M.: J. Antibiot., 70, 435 (2017).
19) Dawson, P. E., Muir, T. W., Clark-Lewis, I., Kent, S. B.: Science, 266, 776 (1994).
20) Johnson, E. C., Kent, S. B.: J. Am. Chem. Soc., 128, 6640 (2006).
21) Shang, S., Tan, Z., Danishefsky, S. J.: Proc. Natl. Acad. Sci. USA, 108, 5986 (2011).
22) Abe, T., Kobayashi, K., Kawamura, S., Sakaguchi, T., Shiiba, K., Kobayashi, M.: J. Gen. Appl. Microbiol. (in press).
![]() 丂
丂