�y�g�s�b�N�X�z
�[�V�R�y�v�`�h�̎����Ǔ��l�H�������n�ɂ�鍇���Z�p�y�����̈��i��╪�q�T���ւ̉��p
������a�A�㓡�C���A���@�T��
����@�E���A����@�E���A����@�E��
�P�D�͂��߂�
���R�E�ɂ����Đ����͔��ɑ��l�ȕ����Y����B�����w�V�R���x�ƌĂ�镨���̒��ɂ́A�����������q���������݂��A���i���邢�͂��̌�╪�q�Ƃ��ČÂ����猤������Ă����B���̒��ł��A���������Y������y�v�`�h���̓V�R���ɂ͑��푽�l�Ȑ����������������̂��m���Ă���A�����q���i�ւ̉��p�����҂���Ă���B�����������������y�v�`�h���V�R���ɂ́A�^���p�N���ɂ͌����Ȃ�����ȕ����\���������̂������B�����͕W�I�����\��זE�����ߐ��A�זE�����萫�Ƃ��������i�ɕK�v�Ƃ���镨���Ɋ�^���Ă��邱�Ƃ��l�����A�y�v�`�h���i���J�������ł�������\���̑��݂����ɏd�v�ł���Ƃ�����B
���̂悤�ȓ���\�������y�v�`�h���A�����͐������n�ƌĂ�镨�����Y�V�X�e����p���āA���Y���Ă���B�������A��ʂɐ������n�ɂ��������ِ��͌����ł��邱�Ƃ������B����y�v�`�h�̐������n�����̗�O�ł͂Ȃ��A�V�R�̓���y�v�`�h�������n�ŗ��p�\�Ȋ���q�͑傫��������B����́A�y�v�`�h���V�R����͕킵���l�H�̃y�v�`�h���J�������ŁA�w���i�Ƃ��Ė��͓I�ȃy�v�`�h���m���Ă���ɂ�������炸�A���̗މ��̂͌���ꂽ��ނ�����邱�Ƃ��ł��Ȃ��x�Ƃ������Ƃ��Ӗ����Ă���A�V�R�̐������n�����̂܂ܗp����헪�ł́A���l�Ȑl�H���i���ƂȂ�y�v�`�h���������邱�Ƃ�����ƌ����悤�B
���̖��̉�����Ƃ��āA��X�͐������Ɋւ��y�f�Q�����A�����������Ǔ��ō������킹�邱�Ƃō\�z����A�č\���^�̎����Ǔ��������n�ɒ��ڂ��A�y�v�`�h�̎����Ǔ������ɉ��p�����B���̕��@�ł́A����ِ�����r�I���e�ȍy�f�����R�ɑg�ݍ��킹�邱�Ƃ��ł����A�e�y�f���l�H�y�v�`�h����������̂ɓK����������T��A�����p���邱�Ƃ��ł���B���ʂƂ��ĉ�X�́A�V�R���ɗގ���������\�������l�H�y�v�`�h���ȕւɍ����ł��鎎���Ǔ��l�H�������n�̍\�z�ɐ��������B�{�e�ł͉�X���J���ɐ��������č\�z�^�̎����Ǔ��l�H�������n�ɂ��Ă��̓������������Ƌ��ɁA�������ՋZ�p�Ƃ������i��╪�q�̒T���Z�p�ɂ��Ă��Љ��B
�Q�D�[�V�R�y�v�`�h�̈��i�ւ̉\��
�y�v�`�h�͐����琔�\���x�̃A�~�m�_���A�Ȃ������q�̑��̂ł���A���̓��ŗl�X�Ȏ�e�̂�y�f�Ƒ��ݍ�p�����邱�Ƃɂ���đ��ʂȐ��������������A�������̐������ۂɊ֗^���邱�Ƃ��m���Ă���B�V�R�ɑ��݂���y�v�`�h�̑����́A20��ނ̃^���p�N�����A�~�m�_����\������钼����̕��q�ł��邪�A���ɂ͎卽N-���`���\���AD-�A�~�m�_�\���A�卽�w�e�����i���i�Ƃ��������ꍜ�i�������̂����݂�1) (�}1)�A���ɍ������������������̂������B�����̓��ꍜ�i��L����y�v�`�h���V�R���́A���������ߐ��E�W�I�F���\�E�v���e�A�[�[����ϐ���L������̂������A���i�Ƃ��Ă̗��p���l�������B�Ⴆ�A�V�N���X�|����A�ƌĂ��V�R���y�v�`�h�́A�Ɖu�}���܂Ƃ��Ĉ�Â̌���ōL���g�p����Ă���2)�B
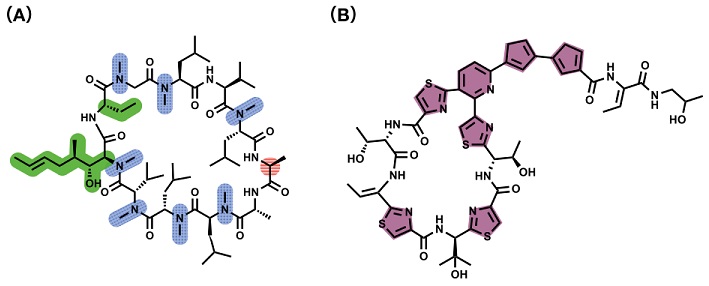

�y�v�`�h���V�R���ɂ݂�����ꍜ�i�𗘗p����A���������ߐ��E�W�I�F���\�E�v���e�A�[�[����ϐ��Ƃ��������i�ɕK�v�ȕ��������˔������l�H�y�v�`�h���J���ł���͂��ł���B�����ʼn�X�́A�V�R���Ɍ��������ȕ����\����g�ݍ��V�K�l�H�y�v�`�h�������Q���w�[�V�R�y�v�`�h�x�ƒ�`�Â�3)�A���̐����������q�T���ւ̉��p��ڎw���Ă���B�[�V�R�y�v�`�h����ՂƂ��������������q�̊J���́A�V�R���₻�̒P���ȍ\���ψّ̂����p����]���̕��@�_�Ɣ�r���āA��蕝�L����܂̑n���ɂȂ���ėp�I�Ȉ��i���J���헪�ƂȂ肤��Ɗ��҂����B
���i�Ƃ��ėL�]�ȋ[�V�R�y�v�`�h��V�K�n������ɂ́A�c��ȑ��l���̋[�V�R�y�v�`�h���C�u�����[�̍\�z�ƒT�������߂��A�������m���Ɗ���e���𗼗�����������@���K�v�ƂȂ�B�`���ɏq�ׂ��ʂ�A�V�R�̐������n�͐����ȉ������������\�ł������ŁA����e�����Ⴍ�A���Y�ł��鉻�����̑��l����������ꍇ�������B�����ʼn�X�́A���ʂȉ������Y�\�Ȑl�H�������n�������Ǔ��ō\�z���邱�ƂŁA���l�ȋ[�V�R�y�v�`�h���C�u�����[�̍������������A����ɂ�������V�K�����������q��T�����邱�Ƃ�ڎw�����B�ȍ~�A��3�߂ł͍č\���^�|��n����ՂƂ����[�V�R�y�v�`�h�̎����Ǔ��l�H�������n�ɂ��āA��4�߂ł͂���ɖ|���C���y�f��g�ݍ��卽�w�e�����i�ܗL�y�v�`�h�����n�ɂ��āA�����đ�5�߂ł͎����Ǔ��l�H�������n�ɂ���č\�z�����[�V�R�y�v�`�h���C�u�����[�̎����Ǔ����q�I���Z�p�ւ̉��p�W�J�ɂ��Ă��ꂼ��Љ��B
�R�D�č\�z�|��n����ՂƂ������ꍜ�i�y�v�`�h�̎����Ǔ�����
�|��n�Ƃ́A�S�Ă̐����ɕ��ՓI�ɑ��݂���^���p�N���������n�ł���B�|��n�ł�mRNA�ɃR�[�h���ꂽ����z��𒒌^�Ƃ��āA����ɑΉ�����A�~�m�_�z������^���p�N�������m�ɍ��������B���̑Ή��Â� (��`�Í��ƌĂ��) �́A�R�h���ƌĂ��mRNA���3�����1�A�~�m�_���Ή����邱�ƂŋK�肳��Ă���B�A�~�m�A�V��tRNA�����y�f (ARS) �������tRNA�ƃA�~�m�_�Ƃ������ɔF�����A���������������邱�ƂŊe�R�h���ɑΉ������A�~�m�A�V��tRNA�����������B���̌����ɐ���E�������ꂽ�A�~�m�A�V��tRNA�����{�\�[���Ɏ�荞�܂�邱�Ƃɂ��AmRNA�ɑΉ������y�v�`�h�E�^���p�N���������ɖ|��Ă���B
���̖|���́A�ւ��y�f��|����q�����ꂼ�ꐸ�����A�����Ǔ��ō������킹�邱�ƂŁA�����Ǔ��ł������ł��邱�Ƃ��m���Ă���4)�B���̂悤�ȍč\�z�^�̖|��n�ł́A�����_���ȉ���z�������mRNA�̍��������������킹�邾���ŁA�������l�����������y�v�`�h���C�u�����[���\�z���邱�Ƃ��ł��邽�߁A�@�\���l�H�y�v�`�h�̑n���Ɋ��p����Ă����B�������A�O�q�̒ʂ�ʏ�̖|��n�ł́AARS�����e����20��ނ̃^���p�N�����A�~�m�_�����p���邱�Ƃ��ł��Ȃ����߁A����╪�q�Ƃ��Ė��͓I�ȓ��ꍜ�i���܂ދ[�V�R�y�v�`�h���C�u�����[�̍\�z�͂ł��Ȃ��B�����ʼn�X�́A�č\���^�̖|��n��l�H�I�ɉ��ς��邱�ƂŁA�[�V�R�y�v�`�h�̍������������鎎���Ǔ��l�H�|��n���m�������B
ARS�������ɃA�~�m�_��F���������ŁA�y�v�`�h���`����S�����{�\�[���͔�r�I��������e���������B�Ⴆ�A�l�H�I�ɒ���������^���p�N�����A�~�m�A�V����tRNA��p���邱�ƂŁA��^���p�N�����̃A�~�m�_���܂ރ^���p�N���̍������\�ł���5)�B��X�́AtRNA�̃A�~�m�A�V�������s�����{�U�C���w�t���L�V�U�C���x���J�����A���푽�l�Ȑl�H�A�~�m�A�V����tRNA�̒�������������6)�B�{��@�ł́A�J���{�L�V����������ꂽ�A�~�m�_�U���̂�tRNA�A�����ăt���L�V�U�C�����������邱�ƂŐl�H�A�~�m�A�V��tRNA������B���̎�@�̗��_�́A�����3�̎���������邾���Ől�H�A�~�m�A�V��tRNA�������\�ł��邽�߁A���Ɋȕւł��邱�ƂƁA�t���L�V�U�C�����l�X�ȃA�~�m�_�U���̂���Ƃ��ċ��e���邽�߁A�ėp�������ɍ������Ƃł���B�e��|����q�̔Z�x���œK�������Ǝ��̎����Ǔ��č\���^�|��n�ɑ��āA���̐l�H�A�~�m�A�V��tRNA��Y�������邱�ƂŁA�e�R�h���Ɋ��蓖�Ă�ꂽ�^���p�N�����A�~�m�_���A���R�ɐl�H�A�~�m�_�ɒu�������� (��`�Í����v���O���~���O) �|�������{���邱�Ƃɐ������� (�}2(A))�B��X�͂��̎����Ǔ��l�H�|��n��FIT (Flexible In vitro Translation) �V�X�e���Ɩ��t���A�[�V�R�y�v�`�h���ȕւ������ɍ��������ՋZ�p�Ƃ��Ċ��p���Ă���7)�B
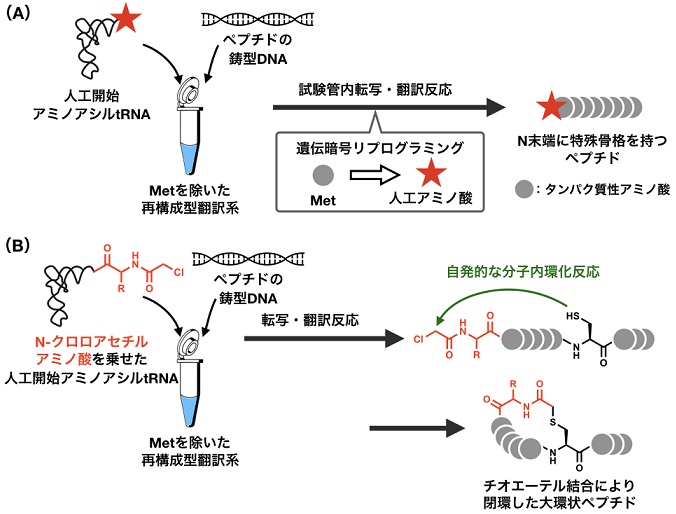
![]()
FIT�V�X�e���ɂ���`�Í����v���O���~���O�̈��Ƃ��āA�|��̊J�n�R�h�������������邱�ƂŁA�y�v�`�h��N���[�ɗl�X�Ȑl�H�I�ȍ\�������邱�Ƃ��\�ł���8)�B�V�R�̐^���ۗR���̖|��n�ł́A���`�I�j��tRNA�����y�f�E���`�I�j��tRNA�z���~���g�����X�t�F���[�[�̓����ɂ��A�z���~�����`�I�j���J�ntRNA����������A���ꂪ�|��̊J�n�R�h��AUG��ǂݎ��B���̌��ʁAN���[�Ƀz���~�����`�I�j�������y�v�`�h�݂̂����������9)�B���FIT�V�X�e���ł́A���`�I�j�����������|��n�t���쐻���A�����ɐl�H�A�~�m�A�V���J�ntRNA�������邱�ƂŁA���`�I�j���̑���ɖ]�݂̃A�~�m�_�ŊJ�n�������s���悤�ɁA�|��n�����݂ɉ��ςł���B�{��@�ɂ�葽�푽�l�Ȑl�H�J�n�A�~�m�_�����������ʁA�|��J�n���������ɕ��L������e�����������Ƃ����炩�ɂȂ����B��̓I�ɂ́A�S�Ẵ^���p�N�����A�~�m�_�⑽�ʂ�N-�A�V���A�~�m�_�A�܂��e��D-�A�~�m�_��Z���y�v�`�h�ł����Ă��J�n�������i�s���A�����̓��ꍜ�i���y�v�`�h��N���[�ɓ����ł��邱�Ƃ������ꂽ8,10,11)�B
�܂��A�J�n�R�h�������łȂ��L���R�h�������������邱�ƂŁA�y�v�`�h�����ɂ����l�ȓ��ꍜ�i�����邱�Ƃ��ł���B�Ⴆ�A�e��N-���`�����A�~�m�_12)�ED-�A�~�m�_13)�Eβ-�A�~�m�_14)�Ȃǂ̗l�X�ȓ��ꍜ�i���܂ރy�v�`�h�̖|�����\�ł���B�܂��A�A�~�m�_�̑���Ƀq�h���L�V�_��A���������邱�ƂŁA�{���|���A�~�h�Y����|��n�Ől�H�I�Ƀ|���G�X�e���������ł��邱�Ƃ������Ă���15)�B�����FIT�V�X�e���œ����ɐ��������r���f�B���O�u���b�N�̒��ɂ́A�]���ł͖|��n�œ����ł��Ȃ��Ƃ���Ă������̂������܂܂��BFIT�V�X�e���̍�������e���́A����������V�R�̃A�~�m�A�V��tRNA�����S�ɏ����ł��Ă��邱�ƁA�܂��|����q�̔Z�x��l�H��ɓK�����Z�x�ɉ��ς��Ă��邱�ƂɗR�����A�����Ǔ��č\���^�̐������n�̒�����[�I�ɕ\���Ă���B
�X��FIT�V�X�e���́A��`�Í����v���O���~���O�ɂ���ċǏ��I�ȓ��ꍜ�i���y�v�`�h���ɓ������邾���łȂ��A�y�v�`�h�S�̂̍��i�����邱�Ƃɂ��������Ă���16)�B���̋�̓I���@��1�Ƃ��ăN�����A�Z�`�����A�~�m�_�ƃV�X�e�C����p�����`�I�G�[�e�����y�v�`�h�̍����@�ɂ��ďЉ��B�{��@�ł́A�J�n�R�h���̈�`�Í����v���O���~���O�ɂ��y�v�`�h��N���[��N-�N�����A�Z�`�����A�~�m�_�����A�y�v�`�h�̉����ɃV�X�e�C�� (Cys) ��z�u���������y�v�`�h��|������B���̌��ʁA�N�����A�Z�`����ƃV�X�e�C���̃`�I�[����|���Ɏ����I�ɔ������A�`�I�G�[�e�������ŕ������y�v�`�h�������� (�}2(B))�B���̔����͕��q���I��I�ɐi�s���A�����|�b�g�Ŋȕւɑ��y�v�`�h�Y�ł���B�܂��A�`�������`�I�G�[�e���ˋ��́A���̓��̃y�v�`�h��^���p�N���Ɍ�����悤�ȃW�X���t�B�h�����Ƃ͈Ⴂ�A���̂̍זE�����̊Ҍ��I�����ł�����ł���17)�B���̐����͐V�K�����������q�̒T����ړI�Ƃ����[�V�R�y�v�`�h���C�u�����[�̍\�z (��q) �ɓK���Ă���B�܂��A���̊���@�͏�q�̐L���R�h���̏��������@�Ƒg�ݍ��킹�邱�ƂŁA���ꍜ�i�����������y�v�`�h�̍����ɉ��p���邱�Ƃ��ł���B
�ȏ�A�č\���^�̖|��n�Ɛl�H�A�~�m�A�V����tRNA��g�ݍ��킹�č\�z���ꂽ�����Ǔ��l�H�|��nFIT�V�X�e���A�y�т��̋Z�p��p���ăy�v�`�h�ɓ������邱�Ƃ̂ł�����ꍜ�i�̗���Љ���BFIT�V�X�e����p���邱�Ƃł���܂ō���������������l�ȋ[�V�R�y�v�`�h�̊ȕւ������ȍ������������A�[�V�R�y�v�`�h�����ɂ�������p�I�Ȋ�ՋZ�p�̊m���ɐ��������Ƃ����悤�B
�S�D�|���C���y�f��p�����卽�w�e���\���ܗL�y�v�`�h�̎����Ǔ�����
FIT�V�X�e����p������`�Í����v���O���~���O�ɂ��AN-���`���\�����i�Ƃ��������l�ȓ��ꍜ�i���y�v�`�h�ɓ������邱�Ƃ��\�ƂȂ������A�V�R���ɂ͑��ɂ����l�Ŗ��͓I�ȓ��ꍜ�i���m���Ă���B�Ⴆ�A�V�R���̃y�v�`�h�̒��ɂ̓A�]�������i��A�]�[�����i�ƌ������卽�w�e���\���������̂����݂���1)�i�}1(B))�B���̎卽�w�e���\���́A�A�~�h���i�Ɣ�r���č\���I�ɍ����ł���a�����������A���ʂƂ��ăy�v�`�h�̕W�I�����\��זE�����ߔ\�ɏd�v�Ȗ������ʂ����Ă���ƍl������B���̂��߁A�卽�w�e���\����L����[�V�R�y�v�`�h�́A���i�Ƃ��č������p���l�����҂ł���B�Ƃ��낪�AFIT�V�X�e�����܂ߒP���Ȗ|���n�ł́A�����I�Ɏ卽�w�e���\�����y�v�`�h���ɓ����ł��Ȃ��B�V�R���y�v�`�h�̐������n�ł́A���{�\�[�����O��̃y�v�`�h��|����ɁA�C���y�f���I���ɍ��i�ϊ����s�����ƂŎ卽�w�e���\�����`������Ⴊ�m���Ă���B��X�͂��̖|���C���y�f��FIT�V�X�e���Ƒg�ݍ��킹�Ċ��p���邱�ƂŁA����ɑ��ʂȍ��i�����y�v�`�h�̎����Ǔ��l�H���������\�ƂȂ�̂ł͂Ȃ����ƒ��z�����B
�����ł͂��̈��Ƃ��āA�V�X�e�C�� (Cys) /�Z���� (Ser) /�X���I�j�� (Thr) �c���Ή�����A�]�������i�ւƕϊ�����E���w�e�����y�fPatD18)�ɂ��ďЉ��BPatD�̓V�A�m�o�N�`���ƌĂ��V�A�m�o�N�e���A�R���̓V�R���y�v�`�h�������n�̖|���C���y�f�ł���B���̍y�f�ɂ��C������O��̃y�v�`�h�ɂ�2�̗̈悪���݂��A�@N���[�̈�Ɉʒu���鋭���ۑ����ꂽ���[�_�[�y�v�`�h (LP) �̈�ƁA�A���̉����Ɉʒu����Cys/Ser/Thr���܂ރR�A�y�v�`�h (CP) �̈悪���݂���BPatD��LP�̈��F�����A���̉����Ɉʒu����Cys/Ser/Thr�c����`�A�]����/�I�L�T�]����/���`���I�L�T�]�������i�ւƕϊ�����y�f�ł���B��X�͂���PatD���E�������AFIT�V�X�e���ɍ������킹�邱�ƂŁAPatD���܂č\���l�H�|��n (FIT-PatD�V�X�e��) ���\�z����19)�B���̌n�ł́A���ꎎ���Ǔ��œ]�ʁE�|��E�A�]�����C���̂R�X�e�b�v�̔����������i�s���邽�߁A�K�Ƀf�U�C����������DNA�f�Ђ��n�t�ɓ���邾���ŁA�����|�b�g�ŃA�]�����ܗL�y�v�`�h�������ł��� (�}3)�B
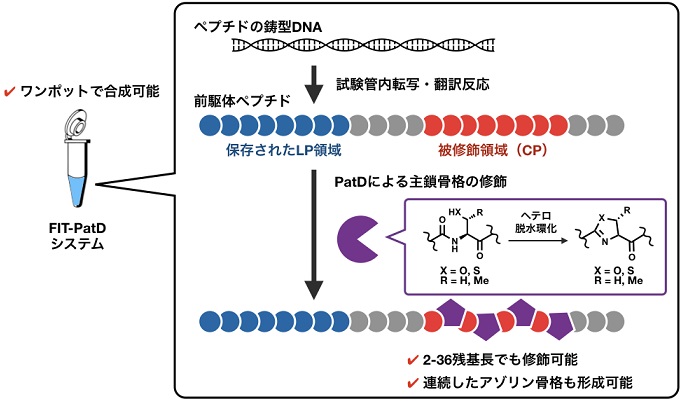
�}3�@FIT-PatD�V�X�e���ɂ��A�]�����ܗL�y�v�`�h�̖|��
�V�A�m�o�N�e���A�ɂ͕�����PatD�̓V�R������݂��Ă��邱�Ƃ���A�{�y�f��������x�̊���e�����������Ƃ͎�������Ă������A���̊���e���̏ڍׁA�܂�ǂ̒��x�̔z��܂ł���Ƃ��Ď������̂��Ƃ������Ƃ�A�ǂ̗l�Ȕz����Ƃ��ċ��e����₷���̂��A�ɂ��Ă͖��炩�ɂȂ��Ă��Ȃ������BPatD�ɂ���ďC������₷���z��̌X�������炩�ɂȂ�A�A�]�����ܗL�[�V�R�y�v�`�h���C�u�����[�\�z�ɖ𗧂m���ƂȂ邱�Ƃ���AFIT-PatD�V�X�e����p���đO��̃y�v�`�h�̊e���ʂɕψق��������푽�l�Ȋ�y�v�`�h�ψّ̂�p�ӂ��APatD�̊���e���������B�O��̃y�v�`�h�ψّ̂��R�[�h�����l�HDNA��FIT-PatD�V�X�e���ɉ����A���̐������ɃA�]�����\�����܂܂�邩�ۂ������ʕ��͂ɂ���Ē�������19,20)�B���̌��ʁAPatD�������z�肳�ꂽ�������ɍ�������e���������A�l�X�Ȑl�H��y�v�`�h��Cys/Ser/Thr�c����A�]�������i�ւƕϊ��ł��邱�Ƃ����炩�ƂȂ����B�Ⴆ�A�V�R�ł�CP��6�`8�c��ł��邪�ACys��1�����܂܂Ȃ�2�c��̒Z��CP��ACys��18���܂�36�c��̒���CP���܂ސl�H��y�v�`�h���C�����邱�Ƃ��ł����B�܂��APatD�͘A������A�]�������i���`�������Ȃ��ƍl�����Ă������A4�̘A������A�]�������i���`���ł��邱�Ƃ��������B�����́APatD���������ėp���y�f�Ƃ��Ă̐��ݐ����AFIT-PatD�V�X�e���̎����Ǔ��č\���������ł��܂����������ƂŁA���������Ǔ�����e�����ł��Ă��邱�Ƃ��������Ă���B�ȏ�A���ʂȃA�]�������i��L����l�H�y�v�`�h�𑽐��������邱�Ƃ��\��FIT-PatD�V�X�e���́A�A�]�����ܗL�[�V�R�y�v�`�h���C�u�����[���\�z���鎎���Ǔ��l�H�������n�Ƃ��č������p�\����L���邱�Ƃ������ꂽ�B
�����ł�FIT-PatD�V�X�e���̕��L������e���̉𖾂Ƃ���𗘗p�������푽�l�ȃA�]�����ܗL�y�v�`�h�̎����Ǔ��������̗���Љ�Ă������APatD�ȊO�ɂ����̑��̖|���C���y�f��p���邱�ƂŁA����ɑ��l�ȓ���\�������y�v�`�h�̍����������I�ɉ\�ł���B���ۂɁAFIT-�V�X�e���ƕ����̃y�v�`�h�C���y�f��g�ݍ��킹�A���i�K�̍y�f�������o�ăA�]�[�����i��E���A�~�m�_�Ƃ����������̓��ꍜ�i��L����[�V�R�y�v�`�h�������Ǔ��Ő��������邱�Ƃɂ��������Ă���21)�B����A�V���Ȗ|���C���y�f��g�ݍ��킹�邱�ƂŁA�����Ǔ��l�H�������n�ō����\�ȓ��ꍜ�i�̃o���G�[�V�������܂��܂������Ă������Ƃ����҂����B
�T�D�����Ǔ��l�H�������n�Ǝ����Ǔ����q�I��@��g�ݍ��킹�����i���T���Z�p
��q������A�̋[�V�R�y�v�`�h�̐l�H�������Z�p�̍ő�̃����b�g�́AmRNA display�ƌĂ�鎎���Ǔ����q�I��@22,23)�Ƒg�ݍ��킹�邱�ƂŁA��K�͂ȋ[�V�R�y�v�`�h���C�u�����[���\�z���A���̒�����]�݂̐���������L����y�v�`�h��v�����ȕւɒT���ł���_�ɂ���B���̎�@�ł�1����ވȏ�Ƃ����c��ȑ��l�������[�V�R�y�v�`�h���C�u�����[���A���g�̃A�~�m�_�z������R�[�h����mRNA�Ń��x�������Ȃ��獇�����邱�Ƃ��\�ł���B����ɂ��A���C�u�����[�̒��������̕W�I�^���p�N���Ɍ�������y�v�`�h�������Ǔ��I�����A�ȕւɋ[�V�R�y�v�`�h���K���h�肷�邱�Ƃ��ł���B
���̈��Ƃ��Ă����ł́A����̑��B�⎩�ȉ��ǐ��nj�Q�Ȃǂ̎����Ɋ֗^����q�X�g���̒E���`�����y�fKDM4A�̍y�f������j�Q����[�V�R�y�v�`�hCP2.3�ɂ��ďЉ��24) (�}4)�BCP2.3�͑��y�v�`�h��O��̂Ƃ��ē���ꂽ�V�K���������[�V�R�y�v�`�h�ł���B�͂��߂ɉ�X��FIT�V�X�e���ɂ��\�z�������y�v�`�h���C�u�����[����AmRNA display��p����KMD4A�Ɍ�������y�v�`�h��T�������B���̌��ʁAKDM4A��KD=29.8 nM�Ƃ������͂Ȍ����e�a���ŃA�C�\�t�H�[���I��I�Ɍ�������ACP2�Ƃ������y�v�`�h�邱�Ƃɐ��������B�������A���̌�̎����ŁA���̃y�v�`�h�������Ǔ��ł�KDM4A�������j�Q���邱�Ƃ����������̂́A�זE���ɂ����Ă͑j�Q�����������Ȃ����Ƃ����炩�ƂȂ����B���̌����Ƃ���CP2�̍זE�����ߐ��ƃy�v�`�_�[�[����ϐ����Ⴉ�������Ƃ��l����ꂽ���߁A��X�͂����̐����̌����ڎw���AKDM4A�Ƃ̌����ɕK�{�łȂ��ƍl�����邢�����̃A�~�m�_��N-���`���\�������A�~�m�_��D-�A�~�m�_�Ƃ���������\�������A�~�m�_�ɒu�������B�������ĉ�X�́A�זE���ł�KDM4A�̑j�Q���������V���ȋ[�V�R�y�v�`�hCP2.3�邱�Ƃɐ��������B���̂��Ƃ́AN-���`���\����D-�A�~�m�_�\���ƌ������[�V�R�y�v�`�h���L�̓���\�����A�]���̃y�v�`�h�ł͒B�������Ȃ��������i�Ƃ��ĕK�v�Ȑ�����t�^���Ă��邱�Ƃ��������Ă���A�[�V�R�y�v�`�h���V�K���i��╪�q�Ƃ��Ĕ��ɍ����|�e���V�������߂Ă��邱�Ƃ�[�I�Ɏ����Ă���ƌ����悤�B
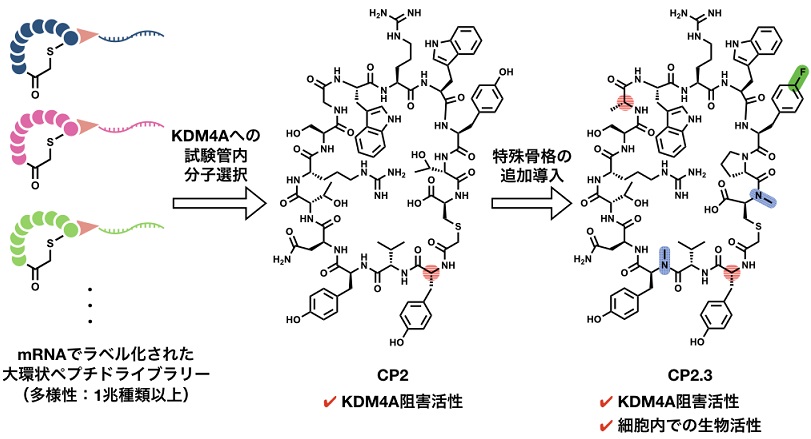

��X�͂��̋[�V�R�y�v�`�h��p���������Ǔ����q�I��@��RaPID (Random non-standard Peptide Integrated Discovery) �V�X�e���Ɩ��t���A���i��╪�q��T������v���b�g�t�H�[���ƂȂ肤���ՋZ�p�Ƃ��ė��p���Ă���B�{��@�̓��M���ׂ������b�g�́A�@���m�̓V�R���ɂ����鐶�������ɗL���ȕ����\����S���V�����y�v�`�h�z��Ƃ����V�K�̑S�̍\���ɑg�ݍ��ނ��Ƃ��ł��邱�ƁA�A�R�̕��̕W�I�����\�Ə����q���̌������萫������ �gdrug-ready�h �Ȓ����q�y�v�`�h��Z���� (�ʏ�2�T�Ԉȓ�) �ɒT���ł��邱�ƁA�����ćB�y�f����זE����e�̂Ɏ���܂ŕ��L�������֘A�^���p�N����W�I�Ƃ��Đݒ�ł��邱�Ƃł���B���ہA����Љ��CP2.3�Ɍ��炸�A���ɑ����̕W�I�^���p�N���ɑ��ċ��������\��j�Q�����������y�v�`�h��RaPID�V�X�e���ɂ���ĒT���E�J������Ă���25-27)�B���݁A�{�Z�p�͎Y�w�A�g�̌`�Ŏ��p����i�߂Ă���Ƌ��ɁA�X�ɑ����̓��ꍜ�i�����[�V�R�y�v�`�h���C�u�����[��p���邱�Ƃ��ł���悤�ɁARaPID�V�X�e�����̂̉��ǂ��i�߂Ă���B�߂������A���܂łɂȂ������[�V�R�y�v�`�h�����i�Ƃ��Ď��X�Ǝ��p������邱�Ƃ����҂ł���B
�U�D������
�ȏ�A�[�V�R�y�v�`�h�̍������\�Ƃ��������Ǔ��l�H�|��n�ł���FIT�V�X�e���ƁA�����ɖ|���C���y�f��g�ݍ��킹�邱�ƂŎ卽���i�̉��ς��\�Ƃ���FIT-PatD�V�X�e���A�X��FIT�V�X�e���ɂ���č\�z�����[�V�R�y�v�`�h���C�u�����[������i��╪�q��T������RaPID�V�X�e�����Љ���BRaPID�V�X�e���͊��ɑ�K�̓��C�u�����[�̍\�z�⊈���[�V�R�y�v�`�h�̑n���̎��p����B�����Ă���A�����̈��i��╪�q�����ݏo����Ă���B���̋Z�p�́A��4�߂ŏЉ���|���C���y�f��FIT�V�X�e����Z���������l�H�������n��g�ݍ��킹�邱�Ƃ��\�ł���A�]���ł͎��������Ȃ��������l�ȋ[�V�R�y�v�`�h�����i��╪�q�̃��C�u�����[�Ƃ��ė��p���邱�Ƃ����҂ł���B�X�ɁA����V���Ȗ|���C���y�f�����������A�����p���č��܂łɂȂ����i�����[�V�R�y�v�`�h�����i���Ƃ��Ď��p���ł���\�����߂Ă���B����͋[�V�R�y�v�`�h���C�u�����[�̂���Ȃ鑽�l���Ɍ����������ƁA��������p�����V�K�����������q�̒T���̑o����W�J�����Ă��������ł���B
�ӎ�
�{�����́A������w��w�@���w�n�����Ȑ��������ɂčs��ꂽ���̂ł��B�{�����Ɋ֘A���������i�߂Ē������w�������ɐS���犴�Ӓv���܂��B�{�����̈ꕔ�́A�Ȍ��� (��茤��A�E����I�G�茤���E�V�w�p�̈挤��)�AJST������������̌��������̂��Ǝ��{�������̂ł���A�����Ɋ��ӂ̈ӂ�\���܂��B
����
1) McIntosh, J. A., Donia, M. S., Schmidt, E. W.: Nat. Prod. Rep., 26, 537 (2009).
2) Nand, K. W., Tim, J. S., Amadeo, J. P., Steven, A. M., Chris, W. C., Roy, M. F.: Ther. Drug Monit., 9, 399 (1987).
3) Goto, Y., Suga, H.: Bull. Chem. Soc. Jpn., 91, 410 (2018).
4) Shimizu, Y., Inoue, A., Tomari, Y., Suzuki, T., Yokogawa, T., Nishikawa, K., Ueda, T.: Nat. Biotechnol., 19, 751 (2001).
5) Xie, J., Schultz, P. G.: Nat. Rev. Mol. Cell Biol., 7, 775 (2006).
6) Murakami, H., Ohta, A., Ashigai, H., Suga, H.: Nat. Methods, 3, 357 (2006).
7) Goto, Y., Katoh, T., Suga, H.: Nat. Protoc., 6, 779 (2011).
8) Goto, Y., Ohta, A., Sako, Y., Yamagishi, Y., Murakami, H., Suga, H.: ACS Chem. Biol., 3, 120 (2008).
9) Meinnel, T., Mechulam, Y., Blanquet, S.: Biochimie, 75, 1061 (1993).
10) Goto, Y., Murakami, H., Suga, H.: RNA, 14, 1390 (2008).
11) Goto, Y., Suga, H.: J. Am. Chem. Soc., 131, 5040 (2009).
12) Kawakami, T., Murakami, H., Suga, H.: Chem. Biol., 15, 32 (2008).
13) Fujino, T., Goto, Y., Suga, H., Murakami, H.: J. Am. Chem. Soc., 135, 1830 (2013).
14) Fujino, T., Goto, Y., Suga, H., Murakami, H.: J. Am. Chem. Soc., 138, 1962 (2016).
15) Ohta, A., Murakami, H., Higashimura, E., Suga, H.: Chem. Biol., 14, 1315 (2007).
16) Iwasaki, K., Goto, Y., Katoh, T., Suga, H.: Org. Biomol. Chem., 10, 5783 (2012).
17) Sako, Y., Goto, Y., Murakami, H., Suga, H.: ACS Chem. Biol., 3, 241 (2008).
18) Schmidt, E. W., Nelson, J. T., Rasko, D. A., Sudek, S., Eisen, J. A., Haygood, M. G., Ravel, J.: Proc. Natl. Acad. Sci. U.S.A., 102, 7315 (2005).
19) Goto, Y., Ito, Y., Kato, Y., Tsunoda, S., Suga, H.: Chem. Biol., 21, 766 (2014).
20) Goto, Y., Suga, H.: Chem. Lett., 45, 1247 (2016).
21) Ozaki, T., Yamashita, K., Goto, Y., Shimomura, M., Hayashi, S., Asamizu, S., Sugai, Y., Ikeda, H., Suga, H., Onaka, H.: Nat. Commun., 8, 1 (2017).
22) Roberts, R. W., Szostak, J. W.: Proc. Natl. Acad. Sci. U.S.A., 94, 12297 (1997).
23) Nemoto, N., Miyamoto-Sato, E., Husimi, Y., Yanagawa, H.: FEBS Lett., 414, 405 (1997).
24) Kawamura, A., Munzel, M., Kojima, T., Yapp, C., Bhushan, B., Goto, Y., Tumber, A., Katoh, T., King, O. N. F., Passioura, T., Walport, L. J., Hatch, S. B., Madden, S., Muller, S., Brennan, P. E., Chowdhury, R., Hopkinson, R. J., Suga, H., Schofield, C. J.: Nat. Commun., 8, (2017).
25) Tanaka, Y., Hipolito, C. J., Maturana, A. D., Ito, K., Kuroda, T., Higuchi, T., Katoh, T., Kato, H. E., Hattori, M., Kumazaki, K., Tsukazaki, T., Ishitani, R., Suga, H., Nureki, O.: Nature, 496, 247 (2013).
26) Hayashi, Y., Morimoto, J., Suga, H.: ACS Chem. Biol., 7, 607 (2012).
27) Morimoto, J., Hayashi, Y., Suga, H.: Angew. Chem. Int. Ed. Engl., 51, 3423 (2012).
![]() �@
�@