【トピックス】
データ駆動型サイエンスによる高次生命現象の理解
青木 航
京大院・農、JST・CREST、JST・創発
1.はじめに
生命現象は極めて複雑である。例えば、代謝・発生・脳機能などは、多数の因子が相互作用することで制御されている。
複雑な生命現象を理解するために、生命科学者はさまざまな方法論を開発してきた。それらの方法論は、「仮説駆動型アプローチ」と「データ駆動型アプローチ」に大別される。仮説駆動型アプローチでは、研究対象となる生命現象を説明可能な仮説を立て、それを検証可能な実験を実施し、当初の仮説が正しかったかどうかを判定する。このアプローチは、合理的に生命現象を解明するための強力な手法である。しかし、関連知識が限定的な場合、もしくは、非常に複雑な生命現象が研究対象となる場合は、うまく仮説を構築できずに機能しないこともある。そのようなケースにおいて、データ駆動型アプローチが強力な手法となる。このアプローチは、研究対象となる生命現象に関連する因子やデータを網羅的に探索・蓄積することで、仮説を生成するための知見を提供する。例えば、cDNAサブトラクション法を用いた本庶らによるPD-1の同定1)や、順遺伝学を用いた大隅らによるオートファジー関連遺伝子の同定2)は、データ駆動型アプローチの強力さを示す好例であろう。
現代において、データ駆動型アプローチは極めて強力な手法となりつつある。ゲノム・トランスクリプトーム・プロテオーム・メタボロームなどの生体関連分子を網羅的に計測するための装置が開発され、また、そこから出力される膨大なデータを解析するための統計学および機械学習的アプローチが整備されてきた (図1)。特に、ゲノミクスとトランスクリプトミクスの発展は著しい。次世代シーケンサー (NGS) とそのデータ解析プラットフォームの発展により、データ駆動型アプローチを誰でも安価に実施できる環境が整いつつある。近年では、細胞の不均一性を計測可能な1細胞NGS解析も実現され、その技術革新のスピードは驚くほどである3)。

一方、プロテオミクスとメタボロミクスにはまだ多くの課題が存在する。プロテオームとメタボロームを分析するためのゴールデンスタンダードは、液体クロマトグラフィーと質量分析計を組み合わせたシステム (LC–MS) である4)。このシステムではまず、生体関連物質混合物を液体クロマトグラフィーにより分離する。分離された各物質は質量分析計に順次導入され、その質量およびフラグメンテーションパターンから物質が同定される。このシステムにより多数の成果が得られてきたが、すべての物質を漏れなく計測することはいまだ難しい。その理由は、①存在量のダイナミックレンジが大きいこと、②すべての物質を捉えられる普遍的な抽出法や分離法がないこと、③各物質のマススペクトルが未整備である点などに由来する。
我々は、上記課題の解決を目指して、モノリステクノロジー5,6)を活用した次世代プロテオミクス・メタボロミクスを開発し、多様な生命現象の動作メカニズムを解明してきた。さらに、分子レベルの従来型オミックス解析の枠を超え、個体レベルの生命現象をデータ駆動で理解するための細胞機能オミックス (機能的セルオミックス) を提唱・確立してきた。本項では、これらの研究成果を概説しつつ、データ駆動型サイエンスの最先端と将来展望を議論したい。
2.モノリステクノロジーによる超高性能
プロテオミクス・メタボロミクスの実現タンパク質と代謝物は核酸のように増幅できない。そのため、少量しかサンプリングできない臨床検体を扱う場合や、微量成分をターゲットとする場合などは分析が難しい。
この状況を改善するためには、クロマトグラフィーの分離性能を向上させることが1つのアプローチとなる。クロマトグラフィーにおける物質分離が不十分だと、多数の物質が同時に溶出されることでイオン化抑制が起き、質量分析計での検出感度が大きく減少する。また、質量だけでは区別できない物質を同定するためにも、高性能な分離が必要となる。そこで我々は、モノリステクノロジーを基盤とした高速液体クロマトグラフィー (high performance liquid chromatography; HPLC) の大幅な改善により、プロテオミクスとメタボロミクスの次世代化を試みてきた。
HPLCは、生体関連物質や医薬品などの精密分離・分析に広く使用されている化学分析法である。一般にHPLCでは、多孔性の化学結合型シリカゲルや有機ポリマーの粒子をステンレス製パイプに均一に充填したカラムが分離媒体として用いられる。HPLCの高性能化は、クロマトグラフィーにおける1つの大きな目標であり、主にGiddingsにより示された式に基づき達成されてきた7)。HPLCが開発された当初は、粒子径10µmの化学結合型シリカゲル充填剤が使用されたが、現在では粒子径5μm程度のものが主流となっている (図2a)。微粒子化は高性能化にとって重要だが、移動相の流路となる粒子間隙を小さくするのでカラム負荷圧の増大を招く。通常のHPLCの十数倍のカラム負荷圧を発生させられる超高速液体クロマトグラフィー (ultra high performance liquid chromatography; UHPLC) において、粒子径1µmの粒子を使用した高分離が達成されているが、特殊な装置を必要とすることから広く普及するには至っていない。
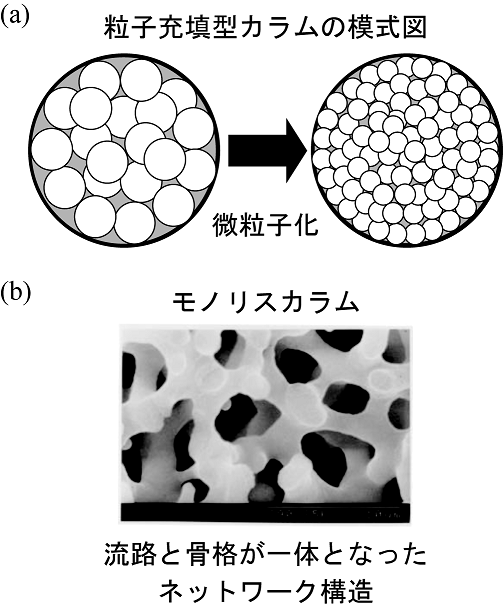

この停滞を打ち破る新しい分離媒体として、モノリスカラムが開発された5,6)(図2b)。モノリスカラムは、流路と骨格が一体となった秩序的なネットワーク構造を有しており、骨格径と流路径をある程度独立して制御できる。そのため、粒子充填型カラムでは不可能であった、細い骨格と大きな流路の同時実現、即ち、高性能分離と低いカラム負荷圧の同時実現が可能となる。
モノリスカラムの特徴を以下に簡単にまとめる。クロマトグラフィーにおける透過率 (permeability) はK値で表され、大きなスルーポアをもつモノリスカラムは一般に粒子充填型カラムより大きなK値を示す。モノリスカラムは大きなK値を示すため、非常に低いカラム負荷圧による送液が可能であり、ロングカラムによる高分離が実現できる。カラムの単位長さあたりのカラム性能を示す理論段高 (H) を指標として見ると、ドメインサイズが小さいモノリスキャピラリーカラムは、同等の線速度で粒子充填カラムと同等以上の性能を示す。総合的なカラム性能を示すセパレーションインピーダンス (separation impedance; E) は単位圧力あたりのカラム性能を表し、モノリスキャピラリーカラムは最適条件下において粒子充填型カラムと同等以上の性能を示す。メートル長の超ロングモノリスカラムを用いることでその分離性能は大きく向上し、複雑な生体関連物質混合物を精密に分離して質量分析計の同定効率を大きく改善できる8-11)。
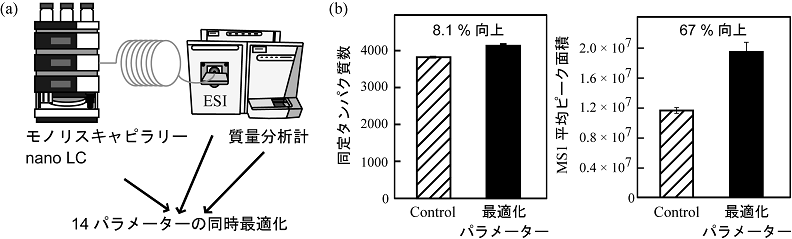
モノリスカラムのポテンシャルをさらに向上させるために、さまざまな観点から改善を試みてきた。第一に、超微量成分を検出可能とするために、モノリスキャピラリーカラムの細径化を試みてきた。LC–ESI–MS/MSにおいて、LCの流量は分析対象物の濃度・イオン化・脱溶媒に大きく影響する。細径化によりLCの流量を減らすと、分析物の濃度が高まるとともに、溶出量が減ることでイオン化や脱溶媒が促進される。そのため、カラムの内径を小さくすることで質量分析計の検出感度が向上する。我々は、モノリスカラムの内径を75µmまで小さくすることでペプチドの検出感度を大きく向上させることに成功し、アトモルオーダーの物質同定をルーチンに実現している12,13)。検出感度を向上させるためには、LC–ESI–MS/MSの各種パラメータを最適化することも重要である。しかし、LC–ESI–MS/MSのパラメータは多数存在し、その最適化には膨大な数の試行が必要とされるため現実的ではなかった。そこで我々は、2因子間交互作用を考慮しつつ各変数の最適値を少ない実験数で決定可能な決定的スクリーニング計画法 (definitive screening design; DSD) を応用した。この統計学的手法により、モノリスnano LC–ESI–MS/MSにおける14種類のパラメータ (LCのグラディエント・ESIの電圧・MSの分解能など) の同時最適化を実現した (図3a)。その結果、最適化前のパラメーターセットを使用した場合と比較して、プロテオミクスアプリケーションにおいて同定タンパク質数を8.1%、MS1平均ピーク面積を67%増加させることに成功した14)(図3b)。

3.モノリステクノロジーを用いた複雑な生命現象の理解
当研究室では、モノリステクノロジーをさまざまな生命現象に適用し、その動作メカニズムの理解を深めてきた。例えば、ALK陽性肺癌のアレクチニブ耐性メカニズムの解明15)、病原性真菌Candida albicansの病原性因子の探索9,10)、植物の二次代謝物質生産メカニズムの解明11)、マメ科植物と根粒菌の根粒形成メカニズムの解明16)などである。本節では、マメ科植物と根粒菌の共生メカニズムに焦点をあて、モノリステクノロジーを用いたデータ駆動型サイエンスのポテンシャルを記述する。
マメ科植物と根粒菌の共生は、最もよく研究されている共生関係の1つである。この共生では、宿主であるマメ科植物に根粒菌が感染し、根に根粒を形成する。この共生関係において、マメ科植物は窒素源を、根粒菌は炭素源と安定的な住処を得る。一方、植物は光合成で得た炭素源の流出と根粒形成のエネルギーというコストを、根粒菌は窒素源の流出というコストを支払う。
この利益とコストの比を最大化するために、根粒は最適な数に保たれる。既知の根粒数制御メカニズムとして、宿主であるマメ科植物が駆動するもの (オートレギュレーション17)と植物ホルモン18)) が知られていた。当研究室では、宿主が駆動するメカニズム以外にも、根粒菌が駆動する根粒数制御メカニズムが存在するのではないかと考え、研究を行った。
根粒菌は2つの生活形態を示す。1つは自由生活状態で、窒素固定を行わない。もう1つは植物と共生するバクテロイド状態で、窒素固定を行う。この2つの形態の根粒菌に対して比較プロテオーム解析を行うことで、根粒形成メカニズムに関わる新規メカニズムを同定できるのではないかと考えた。
従来の方法で根粒菌のプロテオミクスを行う場合、植物から根粒をサンプリングし、そこから根粒菌を単離して、タンパク質を抽出してLC–MS/MSで分析するという手順を踏む必要があった。しかしこのプロセスでは、根粒1,000個 (1 g程度) に相当する大量のサンプルが必要、根粒菌の単離プロセスが複雑なためサンプルがロスする、単離プロセスに半日近くかかるため細胞の状態が変化する、などの問題があった。そこで、従来の1/100にあたる10 mgの根粒サンプルから、植物と根粒菌を分離せずにタンパク質をまとめて抽出し、モノリスnano LC–MS/MSで分析する研究戦略をデザインした。このスキームは、微量かつ複雑なプロテオームを分析する必要があるため難易度が高いが、サンプル量が微量ですむ、サンプルロスが少ない、細胞の状態が変化しにくい、というアドバンテージがある。従来のLC–MS/MSではこのように微量かつ複雑なサンプルの分析は難しかったが、当研究室のモノリスnano LC–MS/MSで分析したところ、根粒菌由来のタンパク質を1,658種類同定することに成功した。この内、共生状態で発現するタンパク質が847種類、自由生活状態で発現するタンパク質が1,533種類であった。これらのタンパク質をゲノムにマッピングしたところ、未知の二次代謝オペロンの発現パターンが共生時に大きく変化することがわかった。
この未知のオペロンの機能を探るために、モノリスカラムを用いたノンターゲットメタボロミクスを実施した。具体的には、この未知オペロンの上流にIPTG誘導性プロモーターを導入した遺伝子組換え根粒菌を構築し、IPTG誘導条件でのみ検出される化合物を同定しようと試みた。IPTG誘導条件でのみ同定された成分は、驚くべきことに、植物ホルモンであるジベレリンであった。なぜ、根粒菌は植物ホルモンであるジベレリンを合成するのだろうか。ジベレリン合成遺伝子を破壊した根粒菌をマメ科植物であるミヤコグサに感染させたところ、接種後3週間までは根粒数が変化しなかったが、接種後4週間から根粒数が有意に増加すること、窒素固定能が減少することが判明した。即ち、根粒菌のジベレリンには、根粒数を抑制し、窒素固定能を上昇させる働きがあることがわかった。この現象の意義は、植物に先に感染した根粒菌がジベレリンを合成することで、更なる根粒菌の感染を阻害し、生存を有利にすることにあると考えられる。この研究は、真正細菌全体で初めてジベレリン合成遺伝子を決定した例となった。
4.個体レベルの生命現象の理解を目指した機能的セルオミックスの提唱
モノリステクノロジーによる次世代プロテオミクス・メタボロミクスは、生体関連分子を網羅的に計測するための非常に強力な技術であるが、うまく適用できない生命現象も存在する。それは、多様な細胞が複雑に相互作用することで創発する個体レベルの生命現象である。例えば、ヒトの脳は1,000億個のニューロンの塊であり、感覚ニューロン・介在ニューロン・運動ニューロン・グリア細胞などが接続しあった複雑なネットワークを形成することで、学習・記憶などの高次機能を創発する。このような複雑な生命現象を理解するためにこそ、データ駆動型アプローチが強力な武器になると期待されるが、分子をターゲットにした従来のオミックス (ゲノミクス・トランスクリプトミクス・プロテオミクス・メタボロミクス) を個体レベルの生命現象にそのまま適用することは難しい。なぜなら、個体中のどの細胞が重要な働きを担っているのか、どの細胞を分子レベルオミックス解析の対象とすべきなのか、は必ずしも自明ではないからである。多数の細胞がネットワークを形成することで創発する個体レベルの生命現象の理解を加速させるためには、それぞれの細胞が生命現象に与える影響を網羅的にアノテーション可能とする新規方法論、即ち、“セルオミックス”と呼べる方法論を確立することが重要であると考えられる。セルオミックスにより、重要な働きをもつ細胞もしくは細胞クラスが同定できれば、分子レベルオミックス解析が適用可能になる。
近年、構造的セルオミックスと呼べるデータ駆動型アプローチが発展しつつある。例えば、ミクロトームもしくは収束イオンビームを電子顕微鏡と組み合わせた連続電子顕微鏡法が開発されており、サブナノメートルの分解能で細胞ネットワークの3D構造をハイスループットに取得可能である19)。また、組織透明化を用いることで、臓器全体から個体全体の細胞ネットワークを迅速に評価可能となりつつある20)。しかし、構造的セルオミックスだけでは細胞ネットワークの“機能”を理解することは難しい。例えば線虫Caenorhabditis elegansにおいては、シドニー・ブレナーらの研究により、302個のニューロンから成る神経ネットワークの全結合パターン (コネクトーム) が既に解明されている21)。この解明から30年以上が経つが、線虫の神経ネットワークと行動の関係はいまだよくわかっていない。
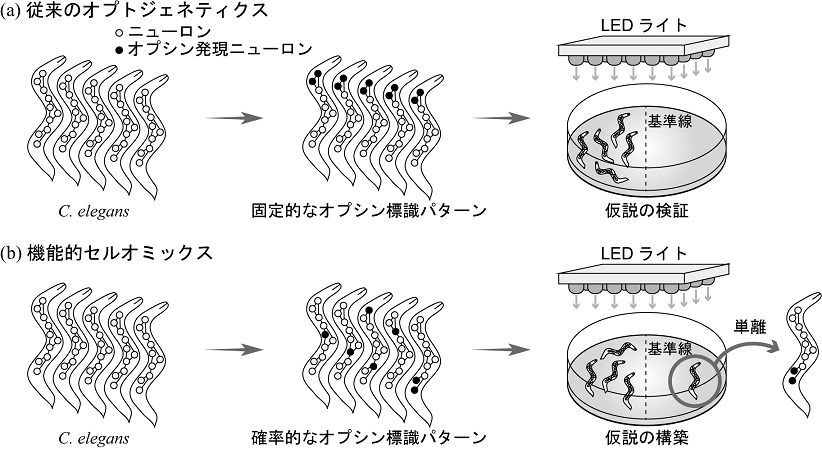
そこで我々は、複雑な神経ネットワークにおいて個々のニューロンが行動に与える影響を網羅的に調べるための方法論―機能的セルオミックス―を提唱してきた22)。機能的セルオミックスにより、個々のニューロンの機能を網羅的にアノテーションすることで、世界で初めて個体レベルのニューラルネットワーク動作モデルを構築できるのではないかと期待される。機能的セルオミックスを実現するためには、データ駆動・ハイスループット・1細胞分解能で、任意のニューロンの活性を自由に制御できればよい。そこで我々は、オプトジェネティクスと確率的Cre/lox部位特異的ゲノム組換えを組み合わせることを着想した。オプトジェネティクスとは、光作動性イオンチャネルであるオプシンをニューロンに発現させ、高い時空間分解能でその活性を自由に制御できる技術である23)。オプトジェネティクスによりニューロンと脳機能の関係を調べられるようになったが、いくつかの技術的弱点があり、神経ネットワークにおける各ニューロンの機能を網羅的に調べるには適していなかった。第一に、オプトジェネティクスは仮説を必要とする。なぜなら、オプシンをニューロンに発現させる際にプロモーターを選択する必要があり、ターゲットとするニューロン群をあらかじめ決定しなければならないからである。第二に、仮説ごとに異なる遺伝子組換え体が要求され、スループットが低いことも挙げられる。第三に、1細胞特異的なプロモーターが非常に少なく、1細胞分解能でニューロンの機能を解析することが難しいこともその理由である。
そこで我々は線虫C. elegansをモデルとして、データ駆動・ハイスループット・1細胞分解能という特徴をもつ新しいオプトジェネティクス (機能的セルオミックス) を開発した (図4)22)。この方法論では、それぞれのニューロンがオプシンで標識されるかどうかを1細胞ごとに確率的に決定することで、オプシン標識パターンがランダム化された線虫個体ライブラリを取得する。この線虫個体ライブラリに対して、LEDライトによる光照射下で行動実験を一斉に行うと、非標準的な行動を示す個体が迅速に単離できる。その個体においてどのニューロンがオプシンを発現していたかを同定すれば、各ニューロンの機能を推定できる。このアプローチは、非標準的な行動を示す線虫を先に単離し、それに関与するニューロンを後から同定するためデータ駆動である。また、無数の標識パターンを一度の実験で検証できるためスループットが高い。さらに、オプシンで標識されるかどうかは1細胞ごとに独立して決定されるため、1細胞分解能の摂動が可能となる。

機能的セルオミックスを線虫に実装するための遺伝子回路を図5aに示した。この遺伝子回路は、4つのプラスミドから構成される。pCreは、ヒートショックに応じて、特定のDNA配列 (lox配列) の間を特異的に組換えることができる酵素Creリコンビナーゼを発現させる。pSTARは、オプシンの確率的標識を実現するための核となるプラスミドであり、全ニューロンで働くプロモーター (F25B3.3p) の下流に、2種類のlox配列 (loxP配列とlox2272配列) を交互に配置し、その間に蛍光タンパク質mCherryと転写因子QF2wをコードする遺伝子を含む。loxP配列とlox2272配列は機能的に直交しており、どちらか片方のみがCreリコンビナーゼによって排他的に組換えられる。pQUAS_ChR2_GFPは、転写因子QF2wに依存してオプシン (チャネルロドプシン2) とGFPの融合タンパク質 (ChR2-GFP) を発現させる。mCherryによる全ニューロンの標識を常に継続させるために、pF25B3.3p_mCherryを用いた。
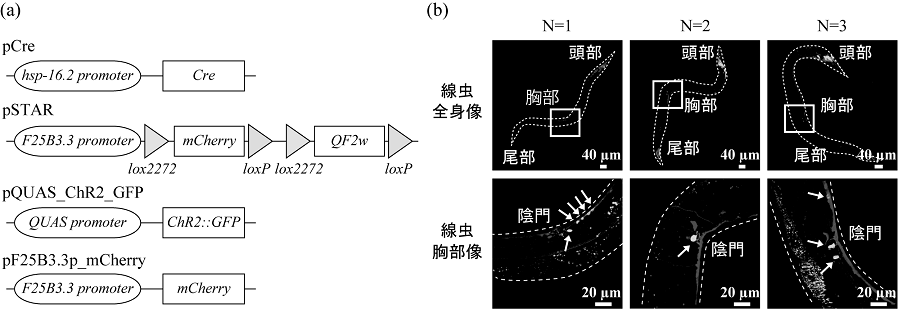

これらのプラスミドを線虫に導入すると、初期状態では全ニューロンでmCherryのみが発現する。ヒートショックによりCreリコンビナーゼを誘導すると、lox2272間の配列が組換えられたニューロンでは転写因子QF2wが発現し、ChR2-GFPが誘導される。実際に、線虫C. elegansにこの遺伝子回路を導入したところ、ChR2-GFPの標識パターンを簡単にランダム化することができた (図5b)。
次に、機能的セルオミックスの実証を目指して線虫の産卵行動に着目した。ChR2-GFPがランダムなパターンで標識された線虫ライブラリに対して光を照射してオプシンを活性化させながら、光依存的な産卵行動を示す個体が現れるかどうかを動画で解析したところ、65%の線虫が光依存的な産卵行動を示した。光依存的に産卵した個体および産卵しなかった個体を共焦点顕微鏡で撮影したところ、産卵個体においてはChR2-GFPがHSN (hermaphrodite-specific neuron; HSN) ニューロンで発現していることがわかった。面白いことに、2つ存在するHSNのうち、片方のみの活性化で産卵行動が誘導されることが判明した。これらの結果は、機能的セルオミックスのコンセプト、即ち、複雑なネットワークを形成する多細胞組織において各細胞の機能をハイスループットにアノテーションできることを実証した成果であると言える。
5.個体をターゲットとした1細胞分解能プロテオミクスの開発
個体における各細胞の機能が明らかになれば、次は、その細胞内部でどのような分子群が働いているのかを調べることが重要である。そこで我々は、個体における特定の細胞のみに対してプロテオミクスを行うための技術を開発してきた。このような実験を可能とするには、目的の細胞のみからタンパク質を特異的に回収する必要がある。そこで我々は、cell-selective BONCAT法 (cell-selective bio-orthogonal non-canonical amino acid tagging) を応用した24,25)。Cell-selective BONCAT法では、phenylalanyl tRNAにアジドフェニルアラニン (Azf) をチャージできる変異型phenylalanyl tRNA syntheatase (MuPheRS) を用いる。Azfに含まれるアジド基はアルキン基と特異的に反応するため、MuPheRSを目的の細胞のみに発現させれば、その細胞に含まれるタンパク質のみをアルキンビーズで濃縮できる。我々は線虫をモデルとして、全ニューロンで働くrab-3プロモーター、もしくは、AFDニューロンのみで働くgcy-8プロモーターを用いて、MuPheRSを発現させた。まず、全神経細胞でMuPheRSを発現させた線虫にAzfを餌として与え、アルキンアガロースを用いてタンパク質を回収し、モノリスnano LC–MS/MSによるプロテオミクスに供したところ、4,412種類のタンパク質を同定することに成功した。同定されたタンパク質には、神経細胞で発現する既知タンパク質が多数含まれていた。同定されたタンパク質のうち、局在パターンが明らかにされていなかったF23B2.10 タンパク質の局在をGFPレポーターアッセイにより確認すると、確かに神経細胞に局在していることが確認できた。これらの結果から、ターゲットとなる細胞にMuPheRSを発現させることで、その細胞のタンパク質のみに対してプロテオミクスを実施できることが示された。続いて、AFDニューロンのみに MuPheRS を発現させた線虫を用いて、同様のプロセスでタンパク質を回収し、モノリスnano LC–MS/MSにより分析した。その結果、1,834種類のタンパク質を同定することに成功し、個体内の単一細胞クラスをターゲットとしたプロテオーム解析を実現した25)。本手法を拡張して個体プロテオームマップを作成できれば、個体レベルの生命現象を理解する上で重要なリソースになると期待される。
6.おわりに
我々はこれまでの研究において、生命現象をデータ駆動で理解するための方法論を多数開発してきた。その成果は、モノリステクノロジーによる超高性能分離を基盤とした次世代プロテオミクス・メタボロミクスや、個体レベルの生命現象を細胞レベルで理解するための機能的セルオミックスとして結実している。
もちろん、まだ課題はある。例えば、質量分析計を用いた従来のプロテオミクスでは、多数の細胞 (数万から数百万細胞) からタンパク質を抽出する必要があり、細胞集団の平均像しか描写できない。細胞の不均一性を計測するためには、1細胞プロテオミクスも必要となってくるだろう。近年、iPAD法26)・OAD法27)・nanoPOTS法28,29)・SCoPE-MS法30,31)・ISPEC法32)など、タンパク質抽出から前処理までをマイクロ・ナノスケールで行いサンプルロスを極限まで減らすことで、1細胞プロテオミクスを実現するサンプル調製法が提案されている。例えばnanoPOTS法では、ナノスケールのウェル中ですべての前処理プロセスを完了できるため、タンパク質の非特異的吸着によるサンプルロスを最小限に抑えられる。nanoPOTS法を高感度なnano LC–MS/MSを組み合わせることで1つのHeLa細胞から1,056のタンパク質の同定が実現されている。
前処理プロセスにおけるサンプルロスの最小化以外にも、LC–MS/MSにおける分離性能やイオン化効率の改善、質量分析計自体の高感度化などにより、微量成分を同定可能とする更なる努力も必要とされている。また、スループットの向上も必要であろう。さらに、従来のオミックスが共通してもつ課題として、1細胞の経時的プロファイリングが難しいことが挙げられる。例えば、細胞を殺さない微小サンプリング手法33)と統合することで、経時的オミックス解析が実現できると期待される。
謝辞
本トピックで紹介した研究成果は、JSPS科研費 (19K08601、17K19452)、JST さきがけ (JPMJPR16F1)、JST CREST (JPMJCR16G2)、JST 創発 (JPMJFR204K) の支援のもと得られたものである。本研究を支えて頂いた植田充美特任教授 (京都大学産官学連携本部)、モノリスカラムを用いたプロテオミクス・メタボロミクスをサポートして頂いた京都モノテックの水口博義博士に心より御礼申し上げる。最後に、LC–MS/MSについて丁寧なご指導を頂いた故森坂裕信助教 (京都大学大学院農学研究科) にこの場を借りて心よりご冥福をお祈りしたい。
文献
1) Ishida, Y., Agata, Y., Shibahara, K., Honjo, T.: EMBO J., 11, 3887 (1992).
2) Ohsumi, Y.: Cell Res., 24, 9 (2014).
3) Hwang, B., Lee, J. H., Bang, D.: Exp. Mol. Med., 50, 1 (2018).
4) Washburn, M. P., Wolters, D., Yates 3rd, J. R.: Nat. Biotechnol., 19, 242 (2001).
5) Minakuchi, H., Nakanishi, K., Soga, N., Ishizuka, N., Tanaka, N.: Anal. Chem., 68, 3498 (1996).
6) Miyamoto, K., Hara, T., Kobayashi, H., Morisaka, H., Tokuda, D., Horie, K., Koduki, K., Makino, S., Núñez, O., Yang, C., Kawabe, T., Ikegami, T., Takubo, H., Ishihama, Y., Tanaka, N.: Anal. Chem., 80, 8741 (2008).
7) Gritti, F., Guiochon, G.: Anal. Chem., 85, 3017 (2013).
8) Yamana, R., Iwasaki, M., Wakabayashi, M., Nakagawa, M., Yamanaka, S., Ishihama, Y.: J. Proteome Res., 12, 214 (2013).
9) Aoki, W., Ueda, T., Tatsukami, Y., Kitahara, N., Morisaka, H., Kuroda, K., Ueda, M.: Pathog. Dis., 67, 67 (2013).
10) Aoki, W., Tatsukami, Y., Kitahara, N., Matsui, K., Morisaka, H., Kuroda, K., Ueda, M.: J. Proteomics, 91, 417 (2013).
11) Takanashi, K., Nakagawa, Y., Aburaya, S., Kaminade, K., Aoki, W., Saida-Munakata, Y., Sugiyama, A., Ueda, M., Yazaki, K.: Plant Cell Physiol., 60, 19 (2019).
12) Ohtani, Y., Aburaya, S., Minakuchi, H., Miura, N., Aoki, W., Ueda, M.: J. Biosci. Bioeng., 128, 379 (2019).
13) Kosaka, Y., Aoki, W., Mori, M., Aburaya, S., Ohtani, Y., Minakuchi, H., Ueda, M.: PLoS One, 15, e0236850 (2020).
14) Aburaya, S., Aoki, W., Minakuchi, H., Ueda, M.: Biosci. Biotechnol. Biochem., 81, 2237 (2017).
15) Tsuji, T., Ozasa, H., Aoki, W., Aburaya, S., Yamamoto-Funazo, T., Furugaki, K., Yoshimura, Y., Yamazoe, M., Ajimizu, H., Yasuda, Y., Nomizo, T., Yoshida, H., Sakamori, Y., Wake, H., Ueda, M., Kim, Y. H., Hirai, T.: Nat. Commun., 11, 74 (2020).
16) Tatsukami, Y., Ueda, M.: Sci. Rep., 6, 27998 (2016).
17) Ferguson, B. J., Indrasumunar, A., Hayashi, S., Lin, M., Lin, Y., Reid, D. E., Gresshoff, P. M.: J. Integr. Plant. Biol., 52, 61 (2010).
18) Ferguson, B. J., Mathesius, U.: J. Chem. Ecol., 40, 770 (2014).
19) Denk, W., Horstmann, H.: PLoS Biol., 2, e329 (2004).
20) Tainaka, K., Kuno, A., Kubota, S. I., Murakami, T., Ueda, H. R.: Annu. Rev. Cell Dev. Biol., 32, 713 (2016).
21) White, J. G., Southgate, E., Thomson, J. N., Brenner, S.: Philos. Trans. R Soc. Lond. B Biol. Sci., 314, 1 (1986).
22) Aoki, W., Matsukura, H., Yamauchi, Y., Yokoyama, H., Hasegawa, K., Shinya, R., Ueda, M.: Sci. Rep., 8, 10380 (2018).
23) Boyden, E. S., Zhang, F., Bamberg, E., Nagel, G., Deisseroth, K.: Nat. Neurosci., 8, 1263 (2005).
24) Yuet, K. P., Doma, M. K., Ngo, J. T., Sweredoski, M. J., Graham, R. L. J., Moradian, A., Hess, S., Schuman, E. M., Sternberg, P. W., Tirrell, D. A.: Proc. Natl. Acad. Sci. USA, 112, 2705 (2015).
25) Aburaya, S., Yamauchi, Y., Hashimoto, T., Minakuchi, H., Aoki, W., Ueda, M.: Sci. Rep., 10, 13840 (2020).
26) Shao, X., Wang, X., Guan, S., Lin, H., Yan, G., Gao, M., Deng, C., Zhang, X.: Anal. Chem., 90, 14003 (2018).
27) Li, Z., Huang, M., Wang, X., Zhu, Y., Li, J., Wong, C. C. L., Fang, Q.: Anal. Chem., 90, 5430 (2018).
28) Zhu, Y., Piehowski, P. D., Zhao, R., Chen, J., Shen, Y., Moore, R. J., Shukla, A. K., Petyuk, V. A., Campbell-Thompson, M., Mathews, C. E., Smith, R. D., Qian, W., Kelly, R. T.: Nat. Commun., 9, 882 (2018).
29) Cong, Y., Motamedchaboki, K., Misal, S. A., Liang, Y., Guise, A. J., Truong, T., Huguet, R., Plowey, E. D., Zhu, Y., Lopez-Ferrer, D., Kelly, R. T.: Chem. Sci., 12, 1001 (2020).
30) Budnik, B., Levy, E., Harmange, G., Slavov, N.: Genome Biol., 19, 161 (2018).
31) Specht, H., Emmott, E., Petelski, A. A., Huffman, R. G., Perlman, D. H., Serra, M., Kharchenko, P., Koller, A., Slavov, N.: Genome Biol., 22, 50 (2021).
32) Hata, K., Izumi, Y., Hara, T., Matsumoto, M., Bamba. T.: Anal. Chem., 92, 2997 (2020).
33) Guillaume-Gentil, O., Grindberg, R. V., Kooger, R., Dorwling-Carter, L., Martinez, V., Ossola, D., Pilhofer, M., Zambelli, T., Vorholt, J. A.: Cell, 166, 506 (2016).
![]()