【トピックス】
SfmC 酵素変換を基盤とした化学-酵素ハイブリッド合成
原口 尚人、谷藤 涼、大栗 博毅
東大院・理
1.はじめに
微生物が生産する“天然物”は、特異な構造に起因した強力な生理活性を発現する。非リボソームペプチド (NRP) は、構造と活性の多様性に富んだ天然物の一群を形成しており、タンパク質性/非タンパク質性アミノ酸から生合成される1,2)。モジュール型酵素である非リボソームペプチド合成酵素 (NRPS) は、主にアミノ酸を縮合してペプチド結合を形成するとともに、ペプチド主鎖や側鎖を改変する多彩な反応を触媒し、多様なNRPを生合成する (図1)。
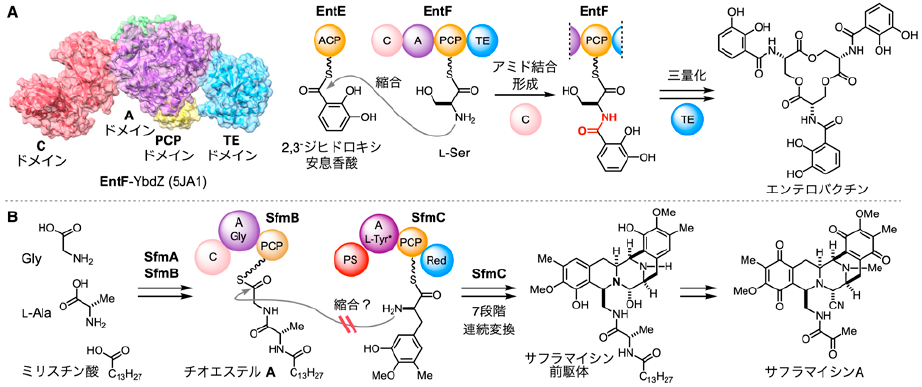

生合成機構とNRPSの立体構造が詳細に研究されている例として、エンテロバクチンが挙げられる (図1A)3-6)。エンテロバクチンの生合成を担うNRPSモジュールEntFは4つのドメイン、C (縮合)、A ( アデニル化)、PCP (ペプチジルキャリアプロテイン)、TE ( チオエステラーゼ) から構成される分子量14万程度のタンパク質である。Aドメインが基質となるL-Serを選択的にアデニル化し、PCPドメイン上にチオエステルとしてロードする。Cドメインが、上流のNRPS モジュールであるEntEにロードされた2,3-ジヒドロキシ安息香酸チオエステルへのL-セリン由来一級アミンの求核攻撃を触媒し、アミド結合を形成する。TEドメインがチオエステルの加水分解と三量化を触媒し、シデロフォアであるエンテロバクチンが生合成される。標準的なNRPSでは、C、A、PCPの3つのドメインの協働的な触媒作用により、アミド結合が順次形成される。これらに加え、EntFのTEドメインに見られるように、マクロ環形成等の特殊な反応を触媒するドメインにより、NRPの骨格多様性が創出される7)。
抗腫瘍性天然物 サフラマイシンAの五環性母骨格は3つのNRPSモジュールによって構築される (図1B)8-11)。標準的なNRPSとして機能する2つの酵素モジュールSfmA、SfmBがミリスチン酸、L-アラニン、グリシンを順次縮合し、SfmBのPCPドメインにチオエステルAを生成する。この下流に位置する特殊なNRPSモジュールSfmCは、L-チロシン誘導体 (Tyr*) とAとのアミド結合形成を触媒せず、特異な7段階連続反応を触媒して高度に官能基化された五環性のサフラマイシン前駆体を一挙に構築する。筆者らは、単純なアミノ酸誘導体から複雑な縮環骨格を簡便かつ迅速に構築するSfmCの環境調和型物質生産システムとしての可能性と有効性に着目し、サフラマイシンAとその類縁体の化学-酵素ハイブリッド合成プロセスを開発している12-16)。本稿では、SfmCの基質適用範囲拡大による非天然型類縁体の合成と、天然物の短段階全合成について紹介する。
2.合成基質の酵素変換:SfmC基質認識
2-1 SfmC推定触媒機構
サフラマイシンAは、放線菌Streptomyces lavendulaeから単離された天然物である (図2)17)。2つのテトラヒドロイソキノリン (THIQ) 環からなる五環性母骨格を有し、強力な抗腫瘍活性を発揮する18)。生産菌が有するSfmCは、分子量約16万の単一酵素 (NRPS) であり、高度に官能基化された母骨格の一挙構築を担う8-11)。まずSfmCは1) ATPを用いてTyr*をアデニル化し、PCPドメイン上にチオエステルとしてロードする。続いて、2) SfmCのC末端に位置するRed (還元) ドメインがSfmB上のチオエステルAを還元し、アルデヒドAとして切り出す。3) PS (Pictet–Spengler) ドメインが、ロードしたTyr*と切り出したアルデヒドAとの位置・立体選択的Pictet–Spengler型環化反応を触媒し、二環性チオエステルBを構築する。4) RedドメインがチオエステルBを還元してアルデヒドBとして切り出しながら、5) AドメインがTyr*を再度ロードする。6) PSドメインが2回目のPS環化を位置・立体選択的に進行させ、四環性チオエステルCを構築する。7) Redドメインがチオエステルを還元してアルデヒドCとして切り出す。アルデヒドCから、二級アミンとアルデヒドとの分子内環化が自発的に進行して五環性骨格が構築され、プレサフラマイシンが生合成される。これが酸化酵素等により修飾を受け、サフラマイシンAが生合成される。SfmAが生合成初期に導入したミリスチン酸は、この後期修飾過程において除去される。一見無駄に思われる脂肪酸の着脱を含んだプロセスであるが、2-2項で後述するように、SfmCによる一連の骨格形成反応において脂肪酸部位は重要な役割を果たしている。
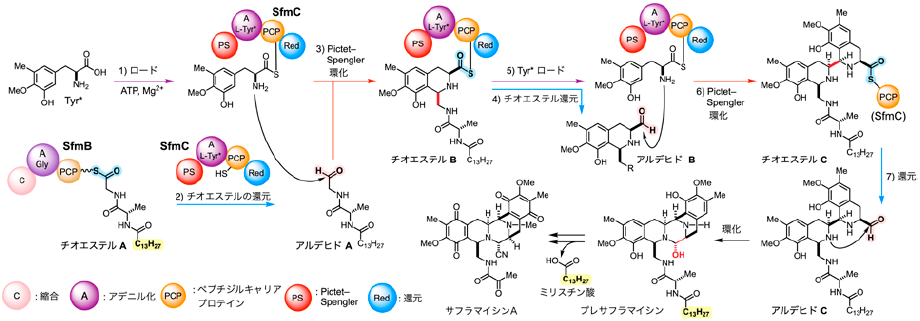
図2 単一NRPSモジュールSfmCによるサフラマイシンA母骨格の生合成機構
2-2 基質構造-SfmC酵素活性相関: 脂肪酸ユニット
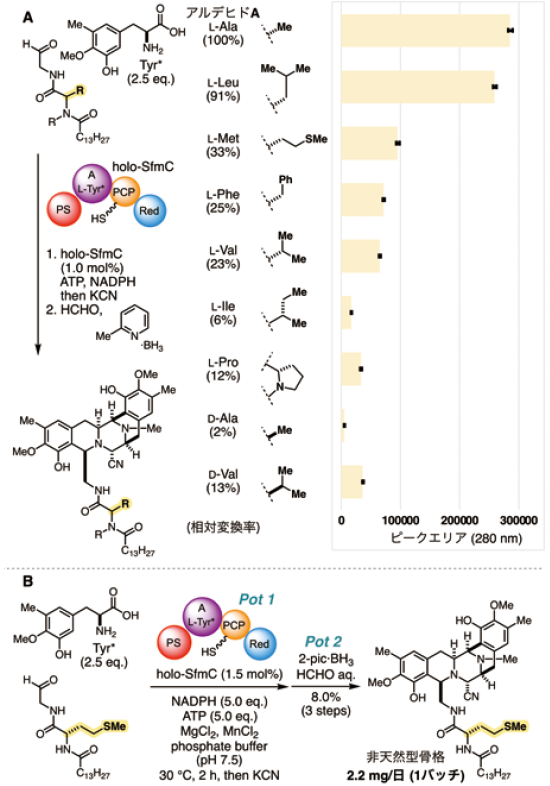
アミノ酸を活性化するNRPSのAドメインは一般的に基質特異性が高く、ゲートキーパーと称される19)。SfmCのAドメインもTyr*を厳密に認識し、L-DOPA等の類縁アミノ酸では、全く酵素変換が進行しない。他方、2-1項の生合成機構を考慮すると、アルデヒドAの結合部位はSfmCのPSドメインと考えられる。アルデヒド型の基質の認識機構は未解明であるが、予備的な実験からPSドメインの基質認識はAドメインに比べて曖昧であることが示唆され、適用範囲の拡大が見込まれた。そこで、アルデヒドAの類縁体を各種化学合成し、SfmCによる変換を試みた。
北海道大学の及川らによる研究から、アルデヒドAの長鎖脂肪酸ユニットの鎖長がSfmCの酵素変換効率に大きな影響を及ぼすことが示唆されていた9)。脂肪酸鎖長と変換効率との相関を定量的に明らかにするため、C12、C14、C16の脂肪酸をそれぞれ縮合した3つのペプチジルアルデヒドを合成した (図3)16)。大腸菌発現系でホスホパンテテニルトランスフェラーゼ Sfpと共発現させ、SfmCをホスホパンテテイン鎖が付加したholo-SfmCとして調製した。別途合成したTyr*12)と3つのアルデヒドから、精製したSfmCを用いて五環性骨格を合成した。反応溶液をLC分析し、五環性骨格由来の280 nmの吸収ピーク面積から酵素変換効率を見積もった。その結果、C14のミリスチン酸を縮合したアルデヒドAが最も効率よく変換されることが分かった。C14のアルデヒドAの変換効率を100%とすると、ラウリン酸・パルミチン酸を縮合したC12・C16のアルデヒドは、それぞれ27%・17%の相対変換率となった。わずか2炭素の違いがSfmCの酵素変換効率を大きく低減させたこととなる。本反応系における脂肪酸側鎖の重要性を明示する結果となった。
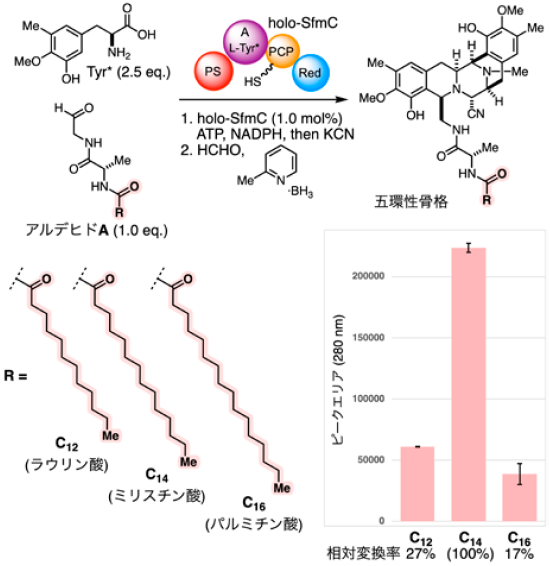
![]()
SfmCと脂肪酸ユニットとの相互作用様式は現時点では明らかになっていない。他方、一連の生合成における脂肪酸の役割の一つとして、生合成産物の自己毒性の低減が考えられる。本生合成プロセスの最終産物であるサフラマイシンAはDNA二重鎖と共有結合を形成し、強力な毒性を発揮する20-22)。一方で、脂肪酸等の長鎖アルキル基を有する合成誘導体はDNAをほとんどアルキル化しない14)。長鎖脂肪酸部位は生合成後期に膜貫通型ペプチダーゼSfmEによって除去され、サフラマイシンAが菌体外へ分泌される23)。これらの知見を統合すると、菌体内では脂肪酸ユニットを導入したプロドラッグとして生合成を進め、然るべきタイミングでDNAアルキル化能を有する生理活性分子へ変換して細胞外へ分泌し、生育環境へ適応するための抗生物質として利用している可能性が示唆される。本機構は、同じ天然物ファミリーであるSF-1739/キノカルシン24,25)、サフラシン26)、エクテナサイジン27,28)群にも保存されている。また、病原性大腸菌が生産するコリバクチン生合成においても同様の機構が提唱されている29)。これらの生合成酵素の一群と脂肪酸ユニットとの相互作用に関する構造解析に基づいた分子認識様式や触媒機構の解明は、今後の意義深い課題と考えられる。
2-3 基質構造-SfmC酵素活性相関:アミド結合
アルデヒドAには、分子中央のL-AlaユニットとN末端側の脂肪酸、C末端側のGlyユニットをつなぐ2つのアミド結合が存在する (図4)。サフラマイシンAのアミドの代わりにエステルが導入された類縁天然物として、レニエラマイシン類が知られている。エステルを導入したアルデヒドをSfmCが変換できれば、類縁天然物の五環性母骨格をワンポットで簡便に合成できると考えられた。
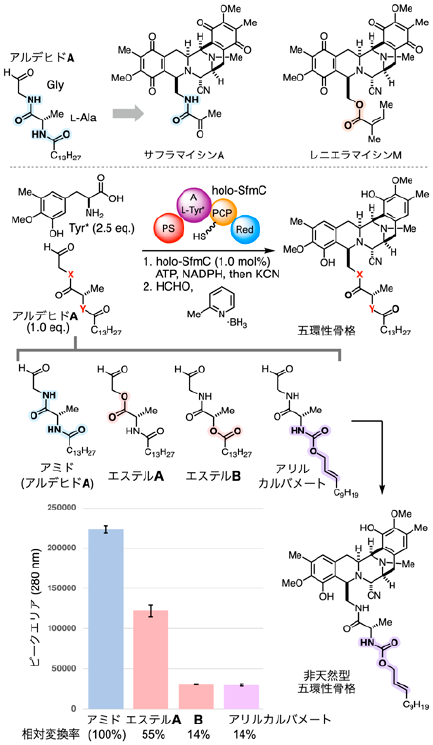
図4 アルデヒドAアミド結合改変アナログとSfmCによる酵素変換
アルデヒドAの2つのアミド結合をそれぞれ改変したエステルA、エステルBを化学合成した (図4)13,16)。また、N末端側のアミドをアリルカルバメートとした基質アナログも設計し、合成した。それぞれTyr*と共にSfmCによる酵素変換を行うと、いずれも対応する五環性骨格へと変換された。2-2項と同様の手順で、SfmCによる相対変換率をそれぞれ算出したところ、C末端側のアミドを改変したエステルAはアルデヒドAの55%程度の相対変換率となった。N末端、即ち、脂肪酸ユニット側のアミドを改変したエステルBとアリルカルバメートは、いずれも14%程度の相対変換率であった。二つのエステルA (55%)、B (14%) の変換率の比較から、SfmCの基質認識において、アルデヒドAのN末端 (脂肪酸) 側のアミド結合周辺の相互作用が重要な役割を果たしている可能性が示唆される。一方、近傍にヘテロ原子や不飽和結合を導入したアリルカルバメート型の基質も酵素変換されることから、更なる基質適用範囲の拡大も見込まれる。SfmCで変換可能なこれらの基質アナログを活用した化学-酵素ハイブリッド全合成については、3項にて後述する。
2-4 基質構造-SfmC酵素活性相関:アミノ酸側鎖
アルデヒドAの2箇所のアミド結合を改変した非天然型合成基質がSfmCに許容されたことから、その間に位置する側鎖 (Me基) についても適用範囲の拡大が期待された。そこで、アルデヒドAのL-Alaを各種L-、D-アミノ酸へと系統的に改変したアナログ群8種を合成した (図5A)16)。これらのアルデヒドとTyr*とのSfmCによる酵素変換を検討したところ、全ての基質アナログから対応する非天然型五環性骨格が得られ、想定以上に基質適用範囲が広いことが分かった。L-Leuを導入したアナログでは天然型のL-Ala導入体とほぼ同等、91%の相対変換率となった。次いでL-Met、L-Phe導入アナログが33%、25%の相対変換率となり、この位置へのヘテロ原子や芳香環の導入すらもSfmCに許容されることが示された。L-Leuよりも小さいL-Val導入体は、予想よりも大幅に低い相対変換率 (23%) となった。L-Leu、L-Met、L-Pheとの比較から、アルデヒドAのペプチド主鎖から1つ以上のメチレン (–CH2–) を有する基質アナログに対して、高い許容能を示すものと考えられる。L-Ile導入体が6%程度に留まっていることもこの作業仮説と合致している。一方、変換効率は低いもののL-ProやD-Ala、D-Val導入アナログからも五環性骨格の生成が確認され、環状アミノ酸や立体化学の反転もある程度許容されることがわかった。
L-Met導入体を用いて、単離スケールでの化学-酵素ハイブリッド合成を実施した (図5B)。SfmCによる酵素変換とKCNの添加によるシアノ化、N-メチル化により、2ポット8%の収率で2.2 mgの非天然型五環性骨格を合成することに成功した。各種生理活性の一次スクリーニングを実施可能なサンプル量を確保しつつ、複雑な天然物類似の化合物を一両日中に供給可能な化学-酵素ハイブリッド合成プロセスを実現することができた。
![]()
3.THIQアルカロイド化学-酵素ハイブリッド全合成
3-1 サフラマイシンA、ジョルナマイシンA
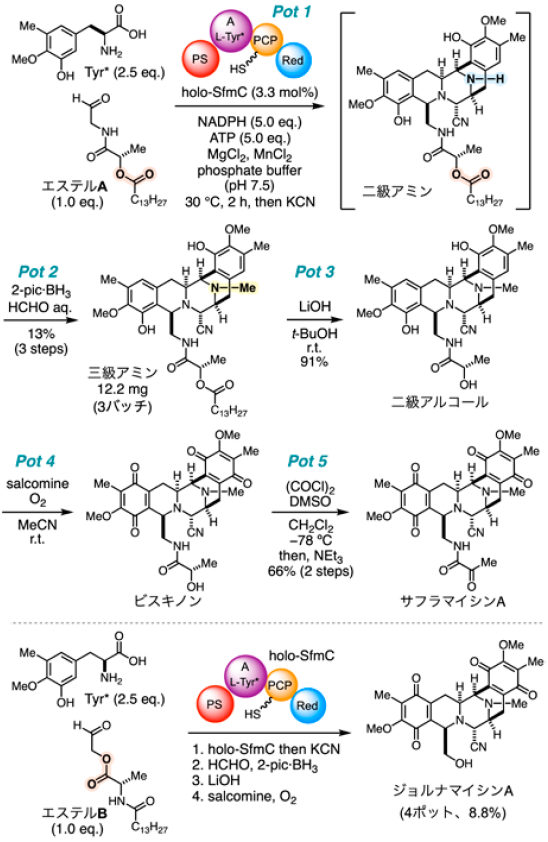
SfmCのin vitro酵素変換と化学変換を融合し、合成基質アナログからTHIQアルカロイド群の化学-酵素ハイブリッド全合成を検討した (図6)13,16)。2-3項で述べたアルデヒドAのアミド結合を改変したエステルAを基質として適用することで、5ポットの変換でサフラマイシンAの全合成を実現した。2.5当量のTyr*と1.0当量のエステルAを3.3 mol%のSfmCにより五環性の二級アミンへ変換した。SfmCを除去した後、ホルムアルデヒドと2-ピコリンボラン (2-pic·BH3) を加えてN-メチル化し、三級アミンとして単離精製した。エステルAから3段階13%程度の収率で五環性骨格を有する三級アミンが得られ、3バッチの合計として12.2 mgを合成することができた。水酸化リチウムを作用させて導入したエステルを加水分解し、二級アルコールを91%の収率で得た。エステルAの適用により、温和な条件下で位置選択的に長鎖脂肪酸ユニットを除去できた。得られた二級アルコールの骨格両端に位置する二つのフェノールをコバルト錯体サルコミンの存在下でキノンへ酸化してビスキノンを合成した。続くアルコールのSwern酸化でα-ジケトンを構築し、目的のサフラマイシンAの全合成を達成した。
![]()
ウミウシJorunna funebrisから単離された類縁天然物ジョルナマイシンAについては、エステルの導入位置が異なるエステルBを基質として、化学−酵素ハイブリッド全合成を検討した。SfmCによる酵素変換・シアノ化からN-メチル化とエステル加水分解、フェノールの酸化を経る僅か4ポットの変換で全合成を実現することができた。
3-2 サフラマイシンY3
サフラマイシンY3は、サフラマイシンAと同様に放線菌Streptomyces lavendulae から単離され、側鎖の末端にアミノ基を有する類縁天然物である39)。アルデヒドAのN末端側アミド結合をアリルカルバメートとした基質アナログを適用し、サフラマイシンY3の化学-酵素ハイブリッド全合成を試みた (図7)13,16)。単離スケールではSfmC酵素変換の効率が向上し、シアノ化とN-メチル化までの3段階で20%の収率、2バッチの合計で12.8 mgの三級アミンが得られた。0価パラジウム触媒とヒドリド還元剤を作用させてアリルカルバメート (紫色ハイライト) を開裂させ、温和な条件下でアルキル基を除去できた。生じた一級アミンを保護し、2段階80%の収率でFmoc保護体を合成した。サルコミンの存在下、酸素酸化で2つのフェノールをキノンとし、N-FmocサラフマイシンY3とした。一級アミンとキノンを有するサフラマイシンY3の単離は困難であったが、末端にアミノ基を有する類縁体の合成を実現できた。SfmC酵素変換に適用する基質アナログを合理的に設計することで、複数の生理活性天然物を迅速に供給できるハイブリッド型合成プラットフォームを構築することができた。
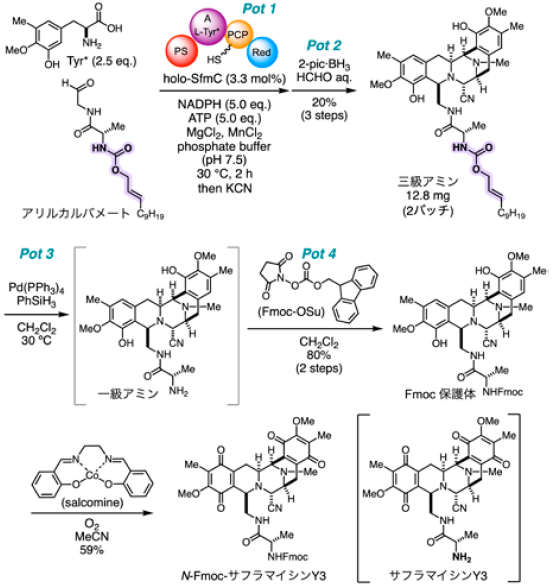
図7 サフラマイシンY3保護体の化学-酵素ハイブリッド合成
4.おわりに
生体分子と特異的に相互作用する生理活性天然物がケミカルツールや医薬品シーズとして重要であることは言を俟たない。縮環骨格に高度に密集した官能基が高い特異性の所以であるが、合成・供給の障壁となるのが常である。NRPSをはじめとする生合成酵素は、天然物の複雑な多環性骨格をほぼ完全な位置・立体選択性にて構築する。一連の多段階カスケードプロセスでは、工程毎の単離精製の手間がなく、保護基も不要である。化学反応では依然として達成困難な連続変換を室温・水中で実現でき、環境調和型の物質変換プロセスとして更なる発展が期待される。
本稿では、放線菌が生産する抗腫瘍性天然物サフラマイシンAの生合成酵素、SfmCを活用した化学−酵素ハイブリッドプロセスについて紹介した。NRPSであるSfmCは、Tyr*とN-アシルジペプチジルアルデヒドの2つのアミノ酸誘導体からサフラマイシンAの五環性母骨格を構築する。2つの基質のうち後者に対して高い基質許容能を示し、アミド結合や構成アミノ酸の改変が可能であった。基質認識が曖昧な酵素は、基質構造の改変により生成物の構造をピンポイントで改変することができる。一つの “catalytic promiscuity” とも言える特性をもつ生合成酵素群は、化学合成との融合により分子構造の多様性を創出でき、創薬リード化合物や機能性分子開発のアプローチとして近年注目されている30)。本研究では基質の構造を系統的に多様化することで、立体構造が未解明な酵素に対する基質特異性や酵素活性と基質構造との相関についての知見を獲得できた。
非天然型基質アナログ群をNRPSモジュールSfmCで変換し、サフラマイシンA、ジョルナマイシンAの迅速で簡便な全合成を実現した。この化学−酵素ハイブリッド合成により、生理活性試験の初期検討が可能なサンプル量の非天然型誘導体を手早く供給できるようになった。酵素を活用した天然物の全合成は、近年続々と報告されている31-37)。生合成酵素工学と有機合成化学との融合は未だ草創期にあり、今後の進展が大いに期待できる。
謝辞
本研究は、著者らが北海道大学及川英秋教授主宰の研究室在籍時に開始したものです。温かくご指導いただきました及川英秋名誉教授、南篤志准教授に深く感謝申し上げます。東京農工大学在籍時に多大な御助力を賜りました、早出広司教授 (現 ノースカロライナ大学チャペルヒル校 教授)、池袋一典教授、津川若子准教授、浅野竜太郎教授、塚越かおり助教に厚く御礼申し上げます。また、JSPS 科研費 (18K19142、19H04647、19H02847、22H00346、22H05127、17J00931、20K15454、22K14790) とJST ACT-X (JPMJAX211A) の支援を受けて実施された研究です。
文献
1) Sussmuth, R. D., Mainz, A.: Angew. Chem. Int. Ed., 56, 3770 (2017).
2) Patel, K. D., MacDonald, M. R., Ahmed, S. F., Singh, J., Gulick, A. M.: Nat. Prod. Rep., 40, 1550 (2023).
3) Gehring, A. M., Bradley, K. A., Walsh, C. T.: Biochemistry, 36, 8495 (1997).
4) Ehmann, D. E., Shaw-Reid, C. A., Losey, H. C., Walsh, C. T.: Proc. Natl. Acad. Sci. USA, 97, 2509 (2000).
5) Drake, E. J., Miller, B. R., Shi, C., Tarrasch, J. T., Sundlov, J. A., Allen, C. L., Skiniotis, G., Aldrich, C. C., Gulick, A. M.: Nature, 529, 235 (2016).
6) Miller, B. R., Drake, E. J., Shi, C., Aldrich, C. C., Gulick, A. M.: J. Biol. Chem., 291, 22559 (2016).
7) Little, R. F., Hertweck, C.: Nat. Prod. Rep., 39, 163 (2022).
8) Li, L., Deng, W., Song, J., Ding, W., Zhao, Q. F., Peng, C., Song, W. W., Tang, G. L., Liu, W.: J. Bacteriol., 190, 251 (2008).
9) Koketsu, K., Watanabe, K., Suda, H., Oguri, H., Oikawa, H.: Nat. Chem. Biol., 6, 408 (2010).
10) Koketsu, K., Minami, A., Watanabe, K., Oguri, H., Oikawa, H.: Curr. Opin. Chem. Biol., 16, 142 (2012).
11) Koketsu, K., Minami, A., Watanabe, K., Oguri, H., Oikawa, H.: Methods Enzymol., 516, 79 (2012).
12) Tanifuji, R., Oguri, H., Koketsu, K., Yoshinaga, Y., Minami, A., Oikawa, H.: Tetrahedron Lett., 57, 623 (2016).
13) Tanifuji, R., Koketsu, K., Takakura, M., Asano, R., Minami, A., Oikawa, H., Oguri, H.: J. Am. Chem. Soc., 140, 10705 (2018).
14) Tanifuji, R., Tsukakoshi, K., Ikebukuro, K., Oikawa, H., Oguri, H.: Bioorg. Med. Chem. Lett., 29, 1807 (2019).
15) Tanifuji, R., Minami, A., Oguri, H., Oikawa, H.: Nat. Prod. Rep., 37, 1098 (2020).
16) Tanifuji, R., Haraguchi, N., Oguri, H.: Tetrahedron Chem, 1, 100010 (2022).
17) Arai, T., Takahashi, K., Kubo, A.: J. Antibiot., 30, 1015 (1977).
18) Scott, J. D., Williams, R. M.: Chem. Rev., 102, 1669 (2002).
19) Stachelhaus, T., Mootz, H. D., Marahiel, M. A.: Chem. Biol., 6, 493 (1999).
20) Lown, J. W., Joshua, A. V., Lee, J. S.: Biochemistry, 21, 419 (1982).
21) Rao, K. E., Lown, J. W.: Chem. Res. Toxicol., 3, 262 (1990).
22) Hill, G. C., Remers, W. A.: J. Med. Chem., 34, 1990 (1991).
23) Song, L. Q., Zhang, Y. Y., Pu, J. Y., Tang, M. C., Peng, C., Tang, G. L.: Angew. Chem. Int. Ed., 56, 9116 (2017).
24) Hiratsuka, T., Koketsu, K., Minami, A., Kaneko, S., Yamazaki, C., Watanabe, K., Oguri, H., Oikawa, H.: Chem. Biol., 20, 1523 (2013).
25) Pu, J. Y., Peng, C., Tang, M. C., Zhang, Y., Guo, J. P., Song, L. Q., Hua, Q., Tang, G. L.: Org. Lett., 15, 3674 (2013).
26) Zhang, Y. Y., Shao, N., Wen, W. H., Tang, G. L.: Org. Lett., 24, 127 (2022).
27) Rath, C. M., Janto, B., Earl, J., Ahmed, A., Hu, F. Z., Hiller, L., Dahlgren, M., Kreft, R., Yu, F., Wolff, J. J., Kweon, H. K., Christiansen, M. A., Hakansson, K., Williams, R. M., Ehrlich, G. D., Sherman, D. H.: ACS Chem. Biol., 6, 1244 (2011).
28) Schofield, M. M., Jain, S., Porat, D., Dick, G. J., Sherman, D. H.: Environ. Microbiol., 17, 3964 (2015).
29) Xue, M., Kim, C. S., Healy, A. R., Wernke, K. M., Wang, Z., Frischling, M. C., Shine, E. E., Wang, W., Herzon, S. B., Crawford, J. M.: Science, 365, eaax2685 (2019).
30) Leveson-Gower, R. B., Mayer, C., Roelfes, G.: Nat. Rev. Chem., 3, 687 (2019).
31) Moore, B. S., Gulder, T. A. M.: Nat. Prod. Rep., 37, 1292 (2020).
32) Li, J., Amatuni, A., Renata, H.: Curr. Opin. Chem. Biol., 55, 111 (2020).
33) Gao, L., Yang, J., Lei, X.: Tetrahedron Chem, 2, 100013 (2022).
34) Vanable, E. P., Habgood, L. G., Patrone, J. D.: Molecules, 27, 6373 (2022).
35) Kaspar, F., Schallmey, A.: Curr. Opin. Biotechnol., 77, 102759 (2022).
36) Paulsel, T. Q., Williams, G. J.: Chembiochem, 24, e202300386 (2023).
37) Li, F., Deng, H., Renata, H.: Nat. Synth., 2, 708 (2023).
38) Charupant, K., Suwanborirux, K., Amnuoypol, S., Saito, E., Kubo, A., Saito, N.: Chem. Pharm. Bull., 55, 81 (2007).
39) Yazawa, K., Takahashi, K., Mikami, Y., Arai, T., Saito, N., Kubo, A.: J. Antibiot., 39, 1639 (1986).
![]()