【トピックス】
酵素内包海洋分解性プラスチックの開発
黄 秋源、木村 聡、岩田 忠久
東大院・農
1.はじめに
近年、廃棄プラスチックによる環境汚染が取り沙汰され、特に海洋では廃棄プラスチックによるゴーストフィッシングやマイクロプラスチック問題が取り上げられている。この問題の解決策のひとつが、環境中に流出した際に水と二酸化炭素にまで完全に分解される生分解性プラスチックへの代替である1) 。これまで様々な生分解性プラスチックが開発されてきたが、次世代の生分解性プラスチックとして求められる性能として、使用中は安定であるが環境中へ流出したら分解が始まる分解開始機能である。
生分解性プラスチックの分解過程は、まず、微生物が分泌する酵素が作用して高分子主鎖骨格が切断され、水に可溶なオリゴマーやモノマーに分解される。続いてそれら低分子が微生物に取り込まれ、微生物体内で二酸化炭素や水、細胞構成物へと代謝される (図1)。最初のステップである生分解性プラスチックの低分子化の過程は微生物が分泌する酵素の作用による分解であることが多いが、非酵素的分解も含まれる。
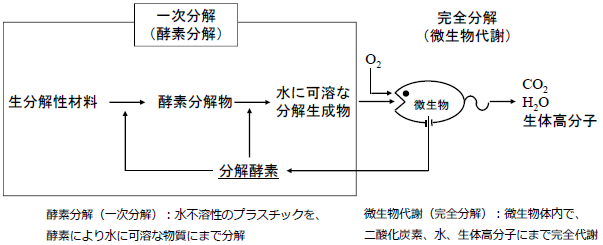
図1 生分解性プラスチックの分解機構:一次分解 (酵素分解) と完全分解 (微生物代謝)
生分解性プラスチックの研究開発は日進月歩であるが、現在最も多く研究開発および生産がなされているのは生分解性脂肪族ポリエステルである。代表的な生分解性ポリエステルとして、微生物産生ポリエステル(PHA)、ポリ乳酸(PLA)、ポリブチレンサクシネート(PBS)、ポリブチレンサクシネートアジペート(PBSA)、ポリカプロラクトン(PCL)、ポリブチレンアジペート-テレフタレート(PBAT)などがある。しかし、PHA、PBSAおよびPCLはあらゆる環境で分解されるものの、これら以外の生分解性プラスチックが分解される環境は限られている。なかでもPLAは、土壌・河川・海洋では決して分解せず、温度60℃以上かつ湿度60%以上のコンポスト環境下でのみ分解されるが、コンポスト中での加水分解は化学反応である。PBS、PBSA、PCLといった生分解性脂肪族ポリエステルはコンポスト環境と土壌中では分解されるが2)、海水中での分解速度は非常に遅い3)。このように、生分解性プラスチックは全ての環境中で容易に分解が進むわけではない。これは、全ての環境に生分解性プラスチックを分解する微生物が豊富に存在しているわけではないこと、つまりは、それぞれの環境中の微生物の種類や量が場所によって異なることに起因している4)。
以上の背景のもと、筆者らは、生分解性プラスチックを分解する微生物が存在しない環境下でも分解が進むように、あらかじめ分解酵素をプラスチック内に埋め込んでおくことを着想した。埋め込まれた酵素は、水が作用しなければ分解作用を起こさないが、プラスチック部材の破断や摩耗によって水がプラスチック内部へ浸透して酵素が活性化され、その時点で生分解が開始されると考えた (図2)。本稿では、市販のクチナーゼを熱混錬により埋め込んだ生分解性ポリエステルの生分解性について概説する5-7)。
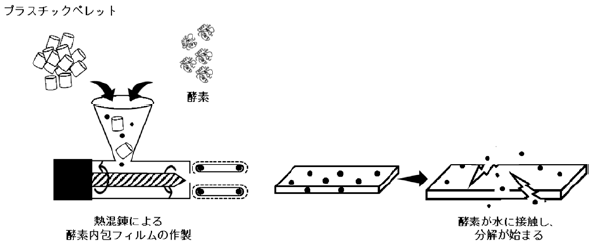
図2 酵素内包生分解性プラスチックの作製と予想される分解様式
2.クチナーゼの基質選択性と熱安定性
2-1 クチナーゼ
酵素は非常に効率のよい生体触媒である。生分解性ポリエステルの分解に関与する分解酵素として、エステラーゼ、リパーゼ、クチナーゼなどがある。これらの分解酵素は、ポリエステルの主鎖構造に存在するエステル結合を加水分解する役割を果たす。クチナーゼ (cutinase) は、主に植物の表面に存在するクチン (cutin) と呼ばれるポリエステルを分解する酵素である。本研究では、市販されている多様なポリエステルを分解できるクチナーゼから、Humicola insolens cutinase (HiC) を選んだ。この酵素を産生するH. insolens は58℃の高温でも増殖できる好熱性真菌であるため8)、HiCは熱安定性が高い。 HiC はα/β-ヒドロラーゼファミリーに属する加水分解酵素であり、触媒トライアド(Ser105、Asp160、および His173) からなる典型的な活性部位を有している9)。3次元構造をみると触媒サイトは表面に露出しており、基質がアクセスしやすい構造をとっている10)。 これにより、HiCは様々なポリエステルへ作用することができる。
2-2 HiCの基質選択性
図3は、酵素水溶液中における様々な市販のポリエステルフィルムの重量減少結果を示している。HiC濃度が100 U/mLの場合、PBAT、ポリD-乳酸(PDLA)、PBS、PBSA、PCLの顕著な重量減少が確認された。図3 (a, b, c) に示すように、各脂肪族ポリエステルフィルムは浸漬の初期段階 (96時間未満) で100%の重量減少を示した。それに対して、芳香族ポリエステルであるPBATは、酵素溶液への96時間の浸漬で64%の重量減少を示した(図2(e))。ポリエチレンフラノエート (PEF) フィルムは、2週間の浸漬で43%の重量減少を示した(図3(f))。一方、ポリエチレンテレフタレート (PET) フィルムは3週間の浸漬後も分解は起こらなかった(図3(g))。過去の研究では、PBAT中のテレフタル酸の含有量が増加すると、酵素加水分解が難しくなることが示されている11)。芳香族ポリエステルの分解性が他の脂肪族ポリエステルよりも低い一因として、剛直な芳香族ポリエステルポリマー鎖は、酵素の活性ポケットに収まりにくいことが原因の一つとして考えられる。また、テレフタル酸または 2,5-フランジカルボン酸を含むポリマー鎖セグメントは硬いのに対し、脂肪族ポリマー鎖セグメントは柔軟である。PEFとポリエチレンテレフタレート (PET) の分解の違いは、ガラス状態のPEFはPET12)より自由体積が高いことから説明できる。
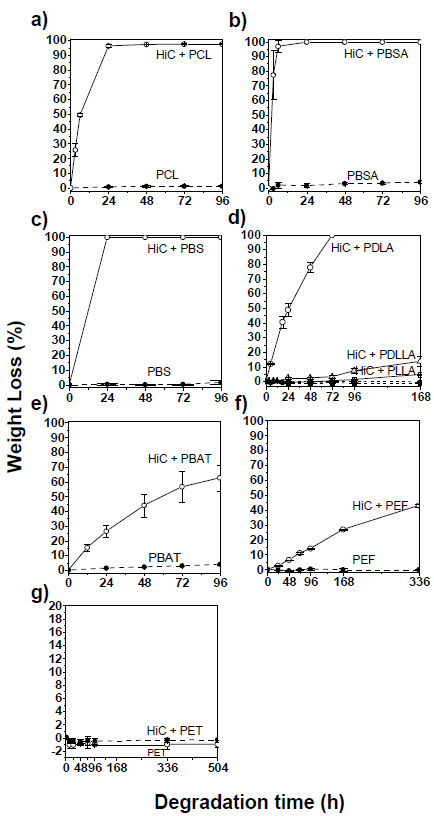

図3(d)は、HiC酵素溶液中でのポリ乳酸の分解速度がポリD-乳酸(PDLA) >> ポリD,L -乳酸(PDLLA) > ポリL-乳酸PLLAの順に遅くなることを示している。ポリ乳酸の分解酵素としては、セリンプロテアーゼ (プロテアーゼKなど) はPLLAを分解するが、PDLAは分解しない13)。一方、α/β-ヒドロラーゼファミリーに属する加水分解酵素 (HiCなど) の触媒トライアドのトポロジーは、セリンプロテアーゼのものとは鏡像であり14)、PLLAとPDLLAよりPDLAを選択的に分解する。
以上の結果より、HiCはポリエステルに対して広い基質選択性を有することを確認できた。分解の速いポリエステルであるPDLA、PBS、PBSA、PCL、PBAT、PEFをHiCを内包させるプラスチックとして選択した。
2-3 HiCの熱安定性
酵素をプラスチックへ内包させる方法としては、溶媒キャスト法と熱混錬法が考えられる。筆者らは、汎用性が高いことと有機溶媒を使用しないことから、熱混錬法による酵素内包を選択した。熱混錬でプラスチックに酵素を内包させるためには、酵素の熱安定性が重要となる。一般的に、水溶液中の酵素は加熱により変性しやすい。これは水が分子潤滑剤として機能することで酵素の立体構造の運動性を高めているためである。すなわち、酵素分子内の疎水性相互作用、静電的相互作用と水素結合で安定化されている酵素タンパク質の立体構造が、高温と水の存在により破壊される。タンパク質の立体構造の安定化に水素結合が寄与している例として、酵素は水溶液中よりも無水有機溶媒中で熱安定性が高いことが報告されている15)。
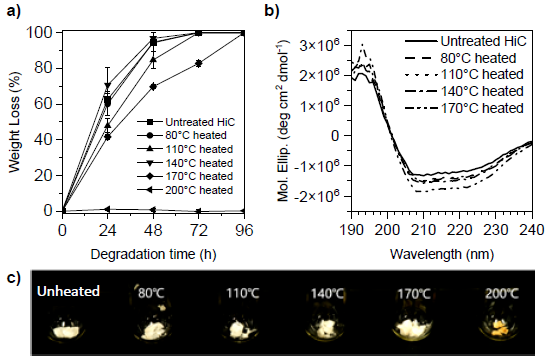
筆者らは、タンパク質の分子内相互作用を破壊しない加熱条件、すなわち無水条件では加熱しても酵素の活性が維持できるのでないかと考え、乾燥した粉末状態の酵素であれば高温での長時間の熱処理に耐えることを発見した。乾燥HiC酵素粉末を10分間熱処理してから調製した酵素溶液にPDLAフィルムを浸漬しながら重量損失をプロットした結果を図4(a)に示す。HiCの活性は140℃以下の加熱ではほとんど変化しないことがわかる。加熱温度170℃は一部が失活しており、200℃では活性が完全に失われた。以上の結果から、酵素は無水条件下ならば加熱処理に対してある程度安定であることがわかった。さらに、図4(c)に示すように、200℃で加熱されたHiC粉末は水に不溶となり、熱誘起凝集が観察された。図4(b)の円二色性スペクトルから、加熱前のHiCと80-170℃で加熱したHiCではスペクトルの変化はわずかであり、HiCは加熱温度170℃までは二次構造を維持できると考えられる。図4(c)に示すように200℃で加熱されたHiC 粉末は変色し、また水に不溶となる熱誘起凝集が観察された。

3.HiC内包ポリエステルの作製と分解挙動
PCL、PBSA、PBS、PBAT、PDLA、PEFのペレットと乾燥HiC粉末 (0.02-0.5 wt%) をそれぞれ、共回転二軸スクリュー押出機で3分間の熱混錬を行った。熱混錬温度は各ポリエステルの融点以上の最低限の加工できる温度に設定された(PCL=90℃、PBSA=100℃、PBS=130℃、PBAT=130℃、PDLA=175℃、PEF=205℃)。その後、混錬押出物を同じ温度で30秒間の熱圧プレスによりフィルムを成形した。
作製したHiC内包PCL、PBSA、PBSフィルム (Hic濃度0.02 wt%) をリン酸緩衝液 (0.1 M、pH7.5)に浸漬し、時間経過に対する重量減少率をプロットすると、いずれも72時間以内に100%の重量減少が観察された(図5(a, b, c))。一方、酵素を内包していないPCL、PBSA、PBSフィルムは緩衝液への浸漬後も重量減少は全く認められなかった。この結果から微量のHicをそのままポリエステルに混入することで、分解性が大幅に向上することがわかった。また、著者らが以前の研究で使用していたCandida antartica lipase B (CaLB)と比較して、HiCの分解活性が極めて高いことも示された6)。図6に分解前後のHiC内包フィルム表面の走査型電子顕微鏡像を示す。分解前のフィルム表面が平滑だったのに対して、分解途中のフィルムの表面には多くの均一な孔構造や凹みが観察された。
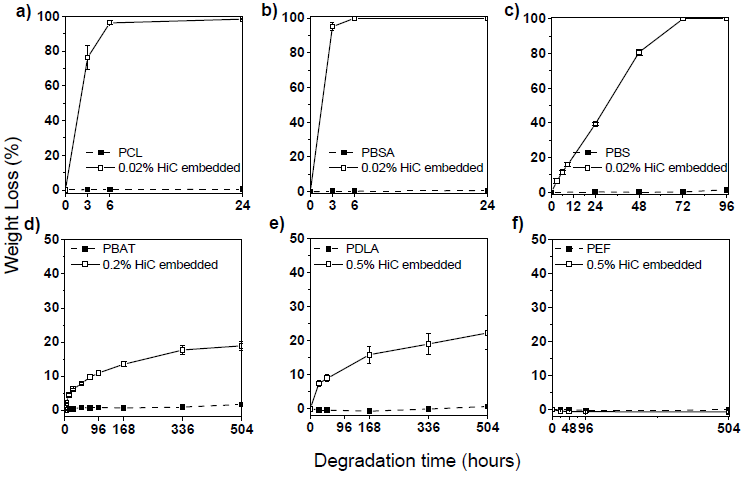
![]()

図6 HiC内包ポリエステルフィルムの分解前及び分解途中の表面形態
HiC内包PBATフィルム (Hic濃度0.2 wt%) では、緩衝液への浸漬3週間後に19%の重量減少が観察された(図5(d))。HiC内包PBATフィルムの重量減少速度は、側鎖のない脂肪族ポリエステルと比較して遅い。上述のように、HiCによる脂肪族芳香族ポリエステルPBATの分解は、酵素水溶液中でも極めて遅い (図1(e))。HiC内包PBATフィルムの重量減少曲線が緩衝液に浸漬後3週間でほぼプラトーに達する理由として、加水分解生成物が酵素分解を阻害している可能性が考えられた16)。
図5(e)に示すように、HiC内包PDLAフィルム (Hic濃度0.5 wt%) では、緩衝液へ浸漬後3週間で22%の重量損失が観察された。酵素を多く内包させてもHiC内包脂肪族ポリエステル (PCL、PBSA、PBS)より分解速度が遅い原因は、PDLAがより分解されにくいことに加えて、175℃の高温加工が酵素に多くダメージを与えたと考えられた。一方、HiC内包PBATとPDLAフィルムの分解途中の様子を走査型電子顕微鏡で観察すると (図6)、HiC内包PBATとPDLAフィルムでは、分解が一様に進行するのではなくサイズの異なる孔構造が発達しながら分解が進行している様子が観察された。分解途中では、直径50µm以下および直径200µm以上の大きな孔構造が観察されたことから、熱混錬過程においてフィルム中で酵素が凝集塊を形成したと考えられた。
図5(f)に、混錬温度205℃でHiCをPEFに内包した結果を示す。HiC内包PEFフィルムは緩衝液に浸漬しても全く分解せず、HiCの熱安定性測定結果(図4(a))と一致した。また、Hic内包PEFフィルムを走査型電子顕微鏡で観察すると、酵素の変性によると思われる直径500µm以上の凝集体が観察された。HiCは粉末状態であっても205℃の高温では立体構造が崩れ、酵素内部に埋もれていた疎水性アミノ酸が露出し、分子間の凝集が促進された可能性がある。
4.クチナーゼ内包ポリエステルの海水中における分解性
海洋環境における HiC内包ポリエステルフィルムの生分解性を評価するため、東京湾から採取した海水を用いて生物化学的酸素要求量 (BOD) 試験を実施した。図7に海水中で25°Cで1ヶ月から2ヶ月間インキュベートしたサンプルの生分解性曲線を示す。標準サンプルであるセルロースは47~65%の最終生分解性を示した。セルロースの最終生分解度の違いは、海水の採取時期、場所、採取時の海水中の微生物数に影響を受ける。酵素を内包していないポリエステルの分解曲線をみると、PBSA、PBS、PBAT、PDLAなどのポリエステルは生分解性プラスチックと呼ばれているが、ほとんどが天然海水中では分解しないか、分解速度が非常に遅いことがわかる。1ヶ月または2ヶ月後でも生分解度は0%のままであった。一方PCLフィルムは時間の経過とともに生分解度が増加した。PCLは天然物であるクチンと構造的に類似しているため、海洋中にも分解に関与する微生物が多く存在していると考えられる17)。
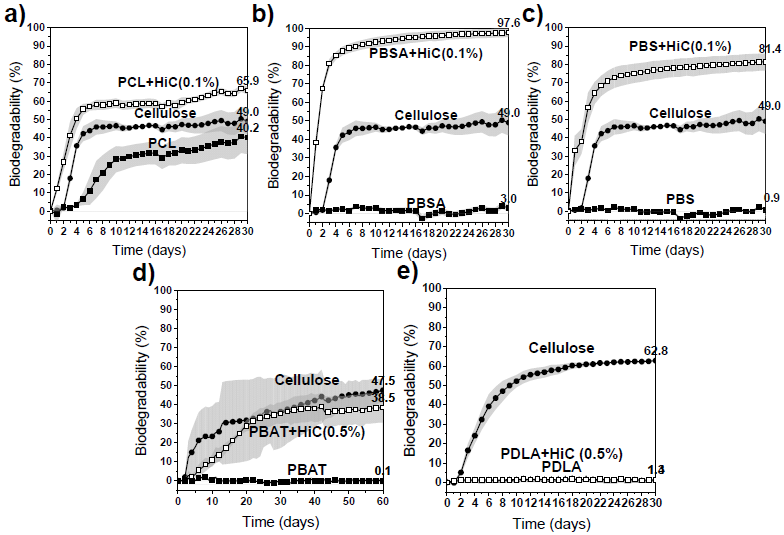
図7 HiC内包ポリエステルフィルムの海水における生物化学的酸素要求量 (BOD) 実験
HiCを内包したフィルムと内包していないフィルムの分解挙動には大きな違いが観察された。酵素内包PCL、PBSA、PBS (HiC濃度0.1 wt%) では、それぞれ66%、98%、および 81%の高い最終生分解度を示した (図7(a, b, c))。さらに、酵素内包PBATフィルムは60日以内にセルロースに近い38.5%の生分解度を示した (図7(d))。酵素内包PBATフィルムの生分解度曲線の初期変化が緩やかなのは、HiCによる加水分解の速度が遅いと考えられる。
図7(e)は、酵素内包PDLAフィルムは2か月後も全く分解しなかったことを示している。酵素内包PDLAフィルムは緩衝液中では重量減少が確認されているので、水可溶性の低分子D-乳酸にまでは分解されていると考えられる (図7(e))。BOD試験の結果は、PDLAの分解生成物は海水中の微生物では代謝できない可能性が考えられ、今後、D-乳酸のモノマー或はオリゴマーのBOD生分解性試験を行うことを計画している。
5.おわりに
市販のクチナーゼHiCをさまざまなポリエステルに内包し、海洋における生分解性を向上させるコンセプトは、実験室レベルでは実証された。酵素内包技術によって、汎用化が遅れている様々な生分解性プラスチックの実用化が可能となり、プラスチックごみ問題の解決への一助となると期待している。酵素内包生分解性プラスチックは、環境流出後に破壊や磨耗により迅速に分解が始まるのでマイクロプラスチックの生成を抑制できると期待される。一方、社会実装のためには、酵素のさらなる耐熱性の向上、分解開始トリガーのさらなる精密化、さらには他の生分解性プラスチックヘの応用など、多くの課題も見えてきている。本研究をさらに発展させ、環境保全に貢献する高性能な生分解性プラスチック部材の開発に貢献したいと考えている。
文献
1) Iwata, T.: Angew. Chem. Int. Ed., 54, 3210 (2015).
2) Bagheri, A. R., Laforsch, C., Greiner, A., Agarwal, S.: Glob. Challenges, 1, 1700048 (2017).
3) Wang, G. X., Huang, D., Ji, J. H., Völker, C., Wurm, F. R.: Adv. Sci., 8, 2001121 (2021).
4) Suzuki, M., Tachibana, Y., Kazahaya, J., Takizawa, R., Muroi, F., Kasuya, K.: J. Polym. Res., 24, 217 (2017).
5) Huang, Q., Hiyama, M., Kabe, T., Kimura, S., Iwata, T.: Biomacromolecules, 21, 3301 (2020).
6) Huang, Q., Kimura, S., Iwata, T.: Polym. Degrad. Stab., 190, 109647 (2021).
7) Huang, Q., Kimura, S., Iwata, T.: Biomacromolecules, 24, 5836 (2023).
8) Ramesh, M., Girish, B., Mahalingeshwara, K. B.: Microbiol. Mol. Biol. Rev., 64, 461 (2000).
9) Kold, D., Dauter, Z., Laustsen, A. K., Brzozowski, A. M., Turkenburg, J. P., Nielsen, A. D., Koldsø, H., Petersen, E., Schiøtt, B., De Maria, L., Wilson, K. S., Svendsen, A., Wimmer, R.: Protein Sci., 23, 1023 (2014).
10) Ferrario, V., Pellis, A., Cespugli, M., Guebitz, G. M., Gardossi, L.: Catalysts, 6, 12, 205 (2016).
11) Müller, R. J., Kleeberg, I., Deckwer, W. D.: J. Biotechnol., 86, 87 (2001).
12) Loos, K., Zhang, R., Pereira, I., Agostinho, B., Hu, H., Maniar, D., Sbirrazzuoli, N., Silvestre, A. J. D., Guigo, N., Sousa, A. F.: Front. Chem., 8, 585 (2020).
13) Reeve, M. S., McCarthy, S. P., Downey, M. J., Gross, R. A.: Macromolecules, 27, 825 (1994).
14) Kawai, F., Nakadai, K., Nishioka, E., Nakajima, H., Ohara, H.: Polym. Degrad. Stab., 96, 1342 (2011).
15) Volkin, D. B., Staubli, A., Langer, R., Klibanov, A. M.: Biotechnol. Bioeng., 37, 843 (1991).
16) de Castro, A. M., Carniel, A., Nicomedes Junior, J., da Conceição Gomes, A., Valoni, É.: J. Ind. Microbiol. Biotechnol., 44, 835 (2017).
17) Suzuki, M., Tachibana, Y., Kasuya, K.: Polym. J., 53, 47 (2021).
![]()